
HCP検出の概念と必要性
生物学的製剤は、通常、宿主細胞(細菌、酵母、哺乳類、昆虫、植物細胞など)での組み換え技術を使用して製造され、モノクローナル抗体、組み換えタンパク質、ワクチンなどが含まれます。これらの製品の製造および精製プロセスでは、宿主細胞の構造タンパク質と宿主細胞から分泌される成長因子の両方を含む宿主細胞タンパク質(HCP)が不純物として必然的に導入され、さまざまな物理化学的および免疫学的特性を持つ複雑な混合物を形成します。高度な精製技術を使用しているにもかかわらず、一部の宿主細胞タンパク質が中間医薬品にプロセス関連の不純物として残っている場合があります。
これらの不純物は潜在的な免疫原性を有し、バイオ医薬品の免疫原性反応を増強し、その安定性と有効性に影響を与え、それによって人体の安全性にリスクをもたらす可能性があります。したがって、HCPを除去するための適切な監視戦略を確立する必要があり、最終製品中のHCP含有量は医薬品の品質要件を満たすように管理する必要があります。
HCPに対する規制要件
生物学的製剤中の HCP の残留含有量は、一般的に製品の重要品質特性 (CQA)、プロセス堅牢性監視の重要な評価指標、および製品の重要な品質管理指標と見なされています。各国の規制当局は HCP に関する規定を設けており、バイオ医薬品の分析と精製により宿主細胞タンパク質 HCP を許容レベルまで低減することを義務付けています。HCP の許容レベルは特定の状況に基づいて評価され、投与量、投与頻度、薬剤の種類、疾患の重症度など、いくつかの要因に依存します。
中国薬局方(2020年版)では、CHO細胞の場合、残留HCPは0.05%未満(500ppm未満に相当)である必要があり、大腸菌の場合、残留HCPは0.01%未満である必要があると規定されています。
米国薬局方 USP <1132> 章では、次のように規定されています。医薬品中の HCP を検出するには、高感度の方法を使用し、含有量は検出限界以下である必要があります (通常 100 ppm 未満、つまり 1 mg の総タンパク質中の HCP 含有量は 100 ng 未満、つまり <0.01%)。
欧州薬局方 EP 2.6.34 では、生物学的製剤では HCP の含有量は 0.1% 未満でなければならないと規定されています。
医薬品規制調和国際会議 (ICH) ガイドライン: ICH Q6B では、残留 HCP を監視するために感度が高く検証済みの方法を採用する必要があり、残留量は通常 100 ppm 未満であることが求められていると規定されています。
規制当局は HCP に対して非常に一貫した要件を持っていることがわかります。生物学的製品のプロセス開発では HCP の検出を実施し、精製プロセスで HCP を安全なレベルまで低減できることを実証する必要があり、最終製品中の残留含有量の推奨制限は中国と米国の両方で 0.01% 未満です。
HCP検出方法
酵素結合免疫吸着測定法(ELISA)は現在、HCP検出に最も一般的に使用されている方法です。2020年版中国薬局方の第3412/3413/3414章に記載されている宿主タンパク質残留物検出法はすべてELISA法です。
米国薬局方 USP <1132> の章では、電気泳動、ELISA、LC-MS/MS などの方法を使用して HCP 含有量を検出できることも記載されており、操作の容易さ、速度、高感度、高スループットのため、HCP 検出の推奨方法として ELISA が推奨されています。
ELISA:
目的: HCP 中のタンパク質の総量を検出し、製品開発やプロセス管理に使用でき、現在 HCP を検出するための一般的な方法です。
利点:高い感度と特異性、高いスループットと自動化機能、定量性、簡単で高速な操作。
欠点:特定の抗体が必要。個々の HCP を具体的に識別して評価できない。方法の開発が複雑で、特別な抗原の準備が必要。方法に影響する要因が多く、HCP の範囲を評価する必要がある。種を識別できない。

ウエスタンブロッティング:
目的:多数のサンプルを一貫してスクリーニングし、抗 HCP 抗体と反応する未知のタンパク質を検出するために使用されます。HCP を検出し、HCP の相対分子量に関するおおよその情報を提供するのに適しています。
利点:特異性。結果は視覚的に直感的: タンパク質のサイズと発現レベルの違いを視覚化します。特定の HCP を検出して定量化できます。
欠点:半定量的。特定のタンパク質に限定され、抗体に結合した HCP のみを検出可能。複雑なタンパク質混合物に対して感度が不十分。タンパク質 SDS 変性により立体配座エピトープが失われる可能性がある。感度はポリクローナル抗体の品質に依存する。
二次元ゲル電気泳動(2-DE):

目的:上流または下流のプロセス開発と特性評価によく使用され、単一のゲル上で異なる HCP を区別できます。
利点:ウェスタンブロッティングが不要で、転送の問題を回避できます。製品から微量の HCP 不純物を分離できます。相対分子量と等電点に関するおおよその情報を提供します。
欠点:タンパク質が多すぎると HCP スポットが隠れてしまう可能性があります。サンプル内の HCP の分布は観察できますが、定量化することはできません。

質量分析:
目的:個々の HCP を識別し、宿主細胞タンパク質の正確な識別および定量情報を提供します。
利点:高解像度と高精度。個々の HCP を識別および監視。HCP の包括的なスペクトルを提供。広範囲のタンパク質をカバー可能。HCP 種を識別可能。
デメリット:高度な設備および技術要件、複雑な操作、高コスト、低いメソッド スループット、複雑なサンプル前処理、潜在的なマトリックス効果、特殊なデータ分析機能が必要。
ELISA は、各国の薬局方により生物製剤中の残留 HCP を検出するために推奨されている方法であり、HCP の総量を測定できます。ただし、HCP の種類と存在量を特定するには限界があり、他の方法で補完する必要があります。各検出方法にはそれぞれ長所と短所があり、実際のアプリケーションでは、実験目的、サンプルの特性、精度、操作の利便性などの要因に基づいて選択し、さまざまな方法を組み合わせて使用することで、より包括的で正確な結果を得ることができます。
さらに、米国薬局方 USP <1132> および欧州薬局方 EP 2.6.34. HOST-CELL PROTEIN ASSAYS では、製品開発のさまざまな段階で HCP 検出に異なる ELISA 試薬を使用する必要があると示されており、HCP 検出方法は市販の試薬、製品/プロセス固有の方法、プラットフォーム方法に分類されています。
USP <1132> では、プラットフォーム メソッドがない場合、市販の試薬を前臨床、フェーズ I、フェーズ II 臨床試験で使用できると記載されています。フェーズ III 臨床試験/プロセス検証および市販後製品では、市販の一般的な HCP 検出試薬の抗体カバレッジが不十分であるなどの制限があるため、細胞の種類やプロセス特異性などの要因を考慮し、上流プロセス開発にはプラットフォーム メソッドまたは製品/プロセス固有のメソッドを使用する必要があります。
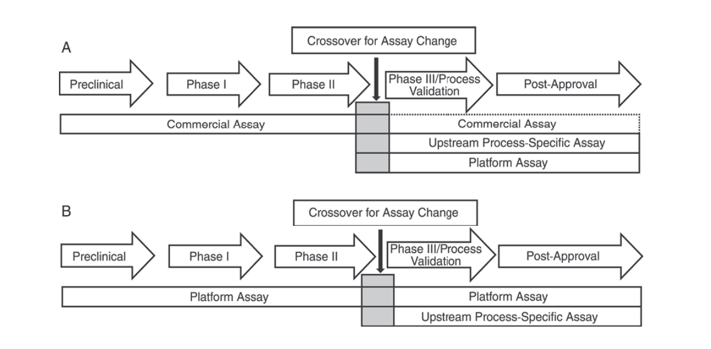
図 1. USP <1132> では、製品開発のさまざまな段階で HCP を検出するための特定の方法を使用することが推奨されています。
|
方法 |
市販試薬キット |
製品/プロセス固有の検出キット |
プラットフォーム検出キット |
|
可用性 |
すぐに入手可能 |
宿主細胞株のクローニングを含む抗原の準備が必要 |
ヌードまたは空ベクター細胞株を使用した抗原の調製が必要 |
|
発達 |
最小限の準備で済む |
抗原特性評価、免疫付与、確認を含む検出の開発には通常少なくとも6か月かかります。 |
抗原特性評価、免疫付与、確認を含む検出の開発には通常少なくとも9か月かかります。 |
|
多様性 |
最も一般的な表現システムに限定 |
様々なタイプの表現システムを使用できる |
様々なタイプの表現システムを使用できる |
|
特異性 |
特異性が低い可能性があり、HCPが検出されない可能性がある |
特異性は通常強化されており、抗体のカバー範囲は特定のHCPパターンをターゲットにしています。 |
市販の検出方法と比較して、特異性が向上し、検出性能の理解が深まり、HCPタイプのカバレッジも増加します。 |
|
料金 |
初期価格は比較的安価だが、下流の精製プロセス全体の開発コストは急速に上昇するだろう。 |
初期コストは高くなりますが、損益分岐点は通常、試薬キット100セットの価値になります。 |
複数の製品に適用できるため、コスト面で大きなメリットがあります。 |
|
依存 |
特定のサプライヤーに依存しているため、試薬や制御スキームは「ブラック ボックス」状態にあります。 |
すべての試薬と緩衝液を包括的に制御し、いつでも調整可能 |
すべての試薬に対する完全な所有権と管理権を持ち、サプライチェーンとバッチ比較におけるリスクを軽減します。 |
|
規制要件 |
販売承認申請のサポートには適していません |
ライセンス製品の初期開発から販売までの全プロセスのサポートに適用可能 |
販売承認までの初期開発支援に適用可能 |
表1. さまざまなHCP試薬キットの比較
Yeasen Biotech カスタマイズ HCP アッセイ開発サービス
HCP 検出の重要性を考慮して、
カスタマイズのタイムライン
宿主細胞残留タンパク質検出キットのカスタマイズサービスには通常 6 ~ 10 か月かかります。
抗原抗体調製: HCP 抗原の決定、HCP 参照標準の確立、免疫経路と戦略の考案、HCP 抗体の調製などには通常 4 ~ 6 か月かかります。
アッセイキットの開発: 抗体の最適化、カバレッジ分析、アッセイキット方法の検証などには通常 2 ~ 4 か月かかります。
Yeasen Biotech HCP 抗体カバレッジ検証サービス
ELISA は業界では残留 HCP を検出するためのゴールド スタンダードであることが知られていますが、この方法の精度は HCP に対するマルチクローン抗体の適用範囲に依存します。米国薬局方と欧州薬局方では、HCP ELISA 抗体とその適用範囲を特徴付けるための推奨方法として、免疫精製と 2D-WB を挙げています。
HCP抗体カバレッジ検証サービス
2D-WB は、まず 2 次元ゲル電気泳動を使用して、サイズと電荷に基づいてタンパク質を分離し、次にこれらのタンパク質を膜に転写して HCP 抗体とインキュベートし、タンパク質インプリントを検出します。
当社は、CHO、大腸菌、HEK293 などの宿主細胞からの HCP サンプルに対する抗体カバレッジ検証サービスを提供するために、2D-WB 機器一式と豊富な実験室経験を備えています。
関連商品:
|
製品 |
カタログNo. | サイズ |
| 36712ES | 48T/96T | |
|
36713ES |
48T/96T | |
|
CHO HCP ELISAキット(CHO-K1) |
36714ES |
48T/96T |
| 大腸菌 HCP ELISA キット (プラスミド) | 36721ES | 48T/96T |