With the deepening research on organoids, an increasing number of individuals are joining in. This article summarizes some common knowledge points about organoids [1-5], hoping to be helpful to everyone.
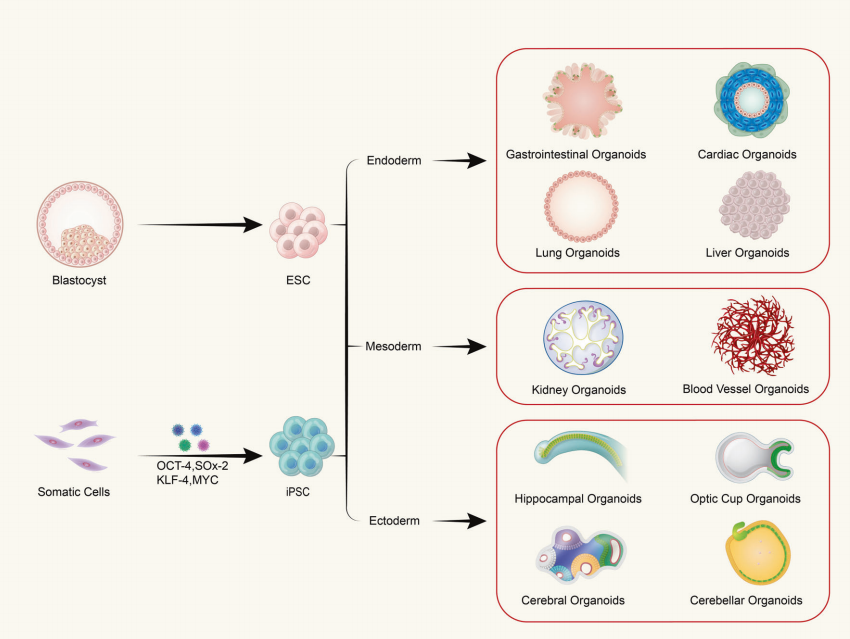
Q: Are organoids composed of a single type of cell or multicellular tissues?
Organoids are formed from the in vitro three-dimensional (3D) cultivation of adult stem cells or pluripotent stem cells, resulting in tissue-like structures with a certain spatial organization. Organoids are not structures composed of single cells; rather, they are formed by inducing division and differentiation of initiating cells with stem cell properties, which then self-assemble into tissues with a certain spatial structure, morphology, and functionality similar to corresponding organs in vivo.
Q: What are the sources for culturing organoids?
(1) Organoids derived from pluripotent stem cells include adult stem cells (ASC), pluripotent stem cells (PSC), and induced pluripotent stem cells (iPSC). (2) Organoids derived from tissue-extracted cells are commonly found in tumor tissues.
Q: Can frozen tissue be used for 3D cultivation in the absence of fresh tissue?
Yes, but there are higher requirements for the size of frozen tissue, and the viability of primary frozen tissue and cells will significantly decrease, leading to a greatly reduced success rate in subsequent cultivation.
Q: How are organoids frozen and resuscitated?
The optimal time to freeze organoids is at passages 2-5, when the activity and differentiation potential of organoids are at their best. The resuscitation of organoids can follow the methods used for cell resuscitation.
Q: Is it necessary to control the size of cultured organoids, and is it beneficial if they are too large?
Yes, it is necessary to control the size, preferably within 500μm, as organoids lack internal vascular and gas-liquid circulation systems. When the size of organoids is large, cells near the center struggle to exchange oxygen and nutrients with the external environment. Therefore, the larger the structure, the greater the number of dead cells.
Q: Besides using matrix gel, what else can be used for culturing organoids?
In addition to matrix gel, alternatives for culturing organoids include (1) decellularized extracellular matrix and other derived proteins, (2) synthetic hydrogels, and (3) engineered recombinant protein gels.
Q: How can directed differentiation of organoids be achieved?
The early development of stem cell-induced differentiation in organoids is jointly regulated by multiple signaling pathways. In vitro cultivation requires the addition of growth factors to simulate the activity of these signaling pathways, guiding cells to differentiate in specific directions. For example, induction with Y27632 and Activin A can differentiate embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) into embryoid bodies (EBs). Subsequently, signaling pathways are regulated by factors such as Wnt3a, FGF-4, and Noggin to induce stem cell differentiation in specific directions.
Q: How can contamination be avoided when obtaining clinical samples?
(1) Ensure aseptic sampling as much as possible. (2) Prior to extraction, immerse in PBS containing antibiotics for several minutes: for tumors located in areas with potential contact with the external environment, such as the stomach, intestines, and bladder, it is recommended to immerse in PBS containing 3%-5% antibiotics for 5-10 minutes; for other common tumors, immerse in PBS containing 1%-2% antibiotics for about 5 minutes. (3) All reagents used during cell extraction should contain 1% antibiotics and an appropriate concentration of primary antibiotics.
Q: What precautions should be taken for the collection, preservation, and transportation of tumor tissues?
Collect tumor tissues with a high content of tumor cells as much as possible, and minimize the exposure time of tissue samples to air to reduce the probability of contamination. Place collected tumor tissue samples into sterile tubes containing special sample preservation solution as soon as possible and transport them rapidly to the testing unit at low temperature (around 4°C) (strive to deliver within 2~4 hours after sampling).
Q: Is there a difference between organoids cultured from lesions and those cultured from adjacent tissues?
What are the requirements for the sampling sites of tumor tissues? Yes, there is a difference. Tumors themselves exhibit heterogeneity, so it's common to observe differences between organoids derived from different sources. Morphologically, organoids derived from primary lesions tend to have a more invasive structure compared to those from adjacent tissues, generally appearing more irregular. To minimize errors in modeling or drug screening, multiple samples should be taken from areas with good activity.
Q: What types of drugs can be used for drug sensitivity testing of tumor organoids?
The main types of anti-tumor drugs in clinical practice can be classified into three categories: cytotoxic drugs (such as paclitaxel, cisplatin/carboplatin, 5-FU, etc.), targeted drugs (drugs targeting EGFR, HER2, VEGFR, etc.), and immunotherapy drugs represented by immune checkpoint inhibitors (PD-1 antibodies, PD-L1 antibodies, etc.).
Q: What is the success rate of PDO cultivation?
The success rate of PDO cultivation varies slightly depending on the source. Most PDOs have a success rate between 63% and 70%, or even higher, up to 90%, which is largely correlated with the activity of the tissue itself. Additionally, clinical treatments may affect the success rate. Success rates can be improved by reducing the time of tissue ex vivo and operational steps.
Q: Can frozen tissue be used for organoid cultivation?
Generally, tissue cryopreservation is not recommended due to significant loss of viability. However, if tissues are stored at -80°C, the optimal window for organoid cultivation is within 6 weeks after preservation. If tissues are stored in liquid nitrogen, the preservation time can be longer but preferably not exceed six months.
Q: When extracting primary cells, there are usually fibroblasts mixed in. How should they be dealt with?
(1) Due to the poor adhesion of fibroblasts, they can be removed by repeated adhesion. (2) Fibroblast removal reagents can be used, but whether they affect organoid cultivation needs experimental validation.
Q: How much original tumor tissue is needed for culturing tumor organoids? Is the biopsy sample sufficient?
Generally, surgical tissues should be larger than the size of 2-3 soybeans; if obtained via needle biopsy, at least 2-3 samples are required, while endoscopic biopsies require at least six or more tumor tissues to be clamped.
Q: If the sample of tumor tissue is too small, and the number of cultured organoids is insufficient for subsequent testing, what should be done?
Since organoids derived from tumor sources may exhibit phenotypic differences after passaging, passaging is generally not recommended. It is recommended in the literature to limit the passaging of organoids to 2-3 generations, with a maximum of 5 generations. If the cell number is too small and cannot meet the testing requirements after 5 generations, consider changing the testing method, such as using a smaller 384-well plate or trying microfluidic chips for testing.
Q: Will there be normal cells in tumor tissues? How to remove these normal cells?
There may be a small number of normal cells. Firstly, try to avoid sampling normal tissues during collection. Secondly, after extracting primary cells, magnetic bead sorting or flow cytometry can be used for further organoid cultivation. When a very small number of normal cells are present, it does not significantly affect subsequent organoid modeling and cultivation, so removal may not be necessary.
Q: When extracting primary cells from tumor tissues, why do the cells appear red?
Tissues are rich in blood supply in vivo, so there are many red blood cells. In most cases, this does not require processing and does not affect organoid cultivation. If there are too many red blood cells, they can be appropriately treated with lysis buffer before cultivation.
Q: During organoid cultivation, black particles are found. How to remove them?
Black particles are most likely impurities or cell debris. They can be removed in two ways:
Digest the organoids and wash them repeatedly with medium to dilute the impurities.
Use a sterile surgical knife to cut the organoids in half, then use a 1ml syringe filled with medium to gently flush out impurities from the organoids.
Q: Is there a limit to the number of passages for organoid cultivation, and how many passages can be conducted?
The number of passages generally depends on the properties of the source cells. Most organoids can be passaged in vitro for up to 10 times (>6 months). The choice of culture conditions may also have some influence, with conditioned medium generally superior to synthetic factor medium.
Q: Can tumor cell lines (such as the HepG2 cell line) be cultured into PDOs?
PDOs are complex self-assembled structures. 3D culture systems formed by single cell lines cannot be called PDOs; they are simply referred to as 3D spherical states.
Q: What are the criteria for passaging organoids?
Depending on the developmental status of the organoids, the time varies, usually between 5-10 days, with a diameter of about 100-200μm. Some slowly developing organoids may take several weeks to reach a suitable passaging state.
Q: How to count the number of viable organoids?
During the experiment, take out the pre-prepared Calcein-AM storage solution and add Calcein-AM solution to the medium to a final concentration of 0.2μmol/L. Incubate at 37°C for 60 minutes. After the time has elapsed, slowly wash away the Calcein-AM-containing medium with PBS and add fresh medium. Use a fluorescence microscope with an excitation wavelength of 490 nm and an emission wavelength of 515 nm to observe and photograph the organoids. Live organoids will appear green and have clear edges. Count the organoids with a diameter >20μm.
Q: How to calculate the viability of organoids?
The viability of organoids is calculated according to the formula: X=(Nlive/Ntotal)×100%, where: X represents the viability of organoids; Nlive represents the number of live organoids; Ntotal represents the total number of organoids.
Q: What are the methods for identifying organoids?
The most basic method is to observe the morphology of organoids through a microscope and perform H&E staining. Further methods include Western Blot, qRT-PCR, immunofluorescence, flow cytometry to detect whether the organoids express corresponding biomarkers. Genetic sequencing can identify the genetic match between the cultured organoids and the source tissue. For some organoids, functional tests can be conducted to see if they possess specific functions. For example, studies have shown that gastric organoids can secrete gastric acid, and cardiac organoids can beat autonomously.
Q: Can normal cells also grow into organoids? How to remove normal organoids during tumor organoid cultivation?
Normal cells can also grow into organoids. Methods to remove normal organoids include: (1) Manual selection based on HE staining results under a microscope; (2) Purification of PDOs by adjusting the composition of the culture medium (such as growth factors/small molecule inhibitors); (3) Dispersing PDOs into single cells for flow cytometry or magnetic bead sorting.
Q: During drug sensitivity experiments, should PDOs be digested from the matrix gel?
No, PDOs need a three-dimensional structure to simulate in vivo conditions. If there is no support from the matrix gel, the accuracy of drug sensitivity experiments will be affected. Generally, soluble drugs can penetrate the matrix gel to act on organoids, but when conducting immunocytochemical experiments, it is necessary to remove the matrix gel.
Q: Can PDO experiments completely replace animal models (PDX)?
PDOs can partially replace PDX, but cannot completely replace them.
Q: What could be the reasons for abnormal growth of PDOs during cultivation, characterized by shortened growth cycles and rapid proliferation compared to previous conditions?
External factors: (1) This abnormality may be caused by the extensive growth of certain contaminating cells, such as fibroblasts. In such cases, it is recommended to perform section staining and observation to confirm the presence of these contaminated cells and then proceed to remove them. (2) Changes in culture conditions, including the addition of certain factors or small molecules, can further activate the proliferation pathways of PDOs.
Internal factors: Possible genetic mutations. To verify this, sequencing is recommended, and the results should be compared with those of the primary PDOs to determine if there are any genetic mutations.
Q: How can the sensitivity of PDOs to drugs be tested?
PDOs can be tested for drug sensitivity using methods such as CCK8 assay, ATP cell viability assay, and live/dead staining. Assessing the ATP activity of tumor organoids is the most common method. ATP is the most important energy molecule in cells and can be used to measure cellular metabolic levels, reflecting the number of viable cells. Based on the effect of drug administration on cellular ATP content, the IC50 value (half-maximal inhibitory concentration of the tested drug) for each drug regimen can be calculated using analysis software to select the most effective drugs for tumor inhibition.
Q: Are the concentration ranges for drug sensitivity experiments of PDOs the same as those for primary tumor cells?
No, they are not the same. Typically, the drug concentration for PDOs needs to be higher than that for primary cells. Preliminary experiments can be conducted to analyze the optimal concentrations for formal drug sensitivity experiments.
Q: At what stage of growth should organoids be used for drug testing?
It is generally recommended to use organoids within 5 passages for drug testing. At this stage, organoids exhibit the best stability and activity.
Q: What are the criteria for determining the success of organoid establishment?
(1) Early preliminary assessment: Organoid morphology changes from a cellular state to forms such as vacuolar, budding, compact, or loose. (2) Identification of specific biomarkers expression, which should be similar to the distribution in tissue slices. Further sequencing analysis can be performed for more detailed comparisons.
Q: How does organoid cultivation differ from regular cell culture?
(1) Different cell culture methods: Organoids require the support of substrates or spatial structures to maintain their three-dimensional structure, while regular cell culture does not require this. (2) Organoid culture requires achieving ex vivo differentiation and self-assembly, thus requiring the use of combinations of various cytokines for induction, resulting in relatively complex culture medium components. Regular cell culture usually involves only a single type of cell, so the culture medium components are relatively simple. (3) Different cell sources: Organoids are derived from multipotent epithelial cells, while regular cell culture is suitable for cultivating various types of selected cells.
Q: How can I determine if the 3D spheres I cultured are organoids and whether they are consistent with the target tissue?
Methods for identifying organoids include H&E staining, immunohistochemistry (IHC), single-cell sequencing, and others. It is necessary to make multidimensional judgments from morphological, histopathological, and molecular genetic perspectives to determine if they are consistent with the target organ or tissue. For tumor organoids, detection of specific biomarkers can be used for confirmation.
Q: If the morphology of the organoids observed during cultivation differs from what is reported in the literature, what could be the reason?
Firstly, individual differences and heterogeneity in sample sources and subtypes may exist. Secondly, differences in the quality of selected cytokines and some small molecule inhibitors used for induction may lead to variations in the differentiation morphology of different organoids. It is suggested to confirm the consistency between organoid morphology and source tissue through methods such as HE staining, IHC, and genetic sequencing, rather than solely relying on literature descriptions.
Q: When conducting drug sensitivity experiments with organoids, is it necessary to control the amount of DMSO used as a solvent for drugs?
Yes, typically drug sensitivity experiments require the volume percentage of DMSO to be less than 0.5%.
Q: How can organoids be recovered from the matrix gel?
The following methods are recommended: (1) Commercially available organoid recovery solutions (CAT#41421ES) can be used to gently and effectively obtain cell suspensions without damaging cells or cell surface proteins. (2) The matrix gel can be thawed at a low temperature to soften it and release the organoids.
Q: Many organoids adhere to the walls of the centrifuge tube during recovery. How can the recovery rate be improved?
When centrifuging after collection, use a horizontal rotor centrifuge and increase the centrifugation speed appropriately. Generally, a centrifugal force of about 300g and a speed of approximately 1000-1200rpm are suitable.
|
Product Name |
CAT |
Size |
| Human Wnt-3a | 92276ES10 | 10μg |
|
92278ES20 |
20μg |
|
|
92701ES10 |
10μg |
|
| Human Noggin | 92528ES10 | 10μg |
|
91330ES10 |
10μg |
|
|
91306ES10 |
10μg |
|
|
91502ES10 |
10μg |
|
|
91701ES08 |
10μg |
|
|
92602ES60 |
100μg |
|
|
91204ES10 |
10μg |
|
|
90601ES10 |
10μg |
|
|
91113ES10 |
10μg |
|
|
92279ES10 |
10μg |
|
|
92055ES10 |
10μg |
|
|
92053ES10 |
10μg |
|
|
92129ES08 |
5μg |
|
|
91304ES10 |
10μg |
|
|
91702ES10 |
10μg |
|
|
92252ES60 |
100μg |
|
|
90103ES10 |
10μg |
|
|
90104ES10 |
10μg |
|
|
90197ES10 |
10μg |
|
|
90144ES08 |
10μg |
|
|
90196ES10 |
10μg |
|
|
90194ES10 |
10μg |
|
|
90111ES10 |
10μg |
|
|
90120ES10 |
10μg |
|
|
90198ES10 |
10μg |
|
|
91605ES10 |
10μg |
|
|
92251ES10 |
10μg |
|
|
92566ES08 |
5μg |
|
|
92102ES10 |
10μg |
|
|
91103ES10 |
10μg |
|
|
92711ES10 |
10μg |
|
|
92122ES60 |
100μg |
|
|
92201ES60 |
100μg |
|
|
92275ES20 |
20μg |
|
|
Human BMP-2 |
92051ES10 |
10μg |
Related reading:
Reference