High-throughput sequencing, often referred to as next-generation sequencing (NGS) technology, represents a significant leap forward from the initial DNA sequencing methods, such as Sanger sequencing. NGS allows for the simultaneous profiling of hundreds of thousands, if not millions, of nucleic acid molecule sequences. Its merits include exceptional throughput, cost-effectiveness, scalability, and a broad spectrum of applications, establishing it as the predominant sequencing technology worldwide.
The NGS sequencing workflow encompasses four primary phases: sample preparation, library construction, sequencing, and data analysis. Central to library construction is the attachment of standardized NGS platform adapter sequences to both ends of fragmented genomic DNA. This step aims to generate an ample supply of library nucleic acid molecules, prepared for sequencing on the NGS instrument through PCR amplification. Depending on the nature of the sample, NGS library construction can be categorized into DNA library construction and RNA library construction. Enzymes play a pivotal role in these interconnected experiments. So, which key enzymes are involved in the process of library construction?
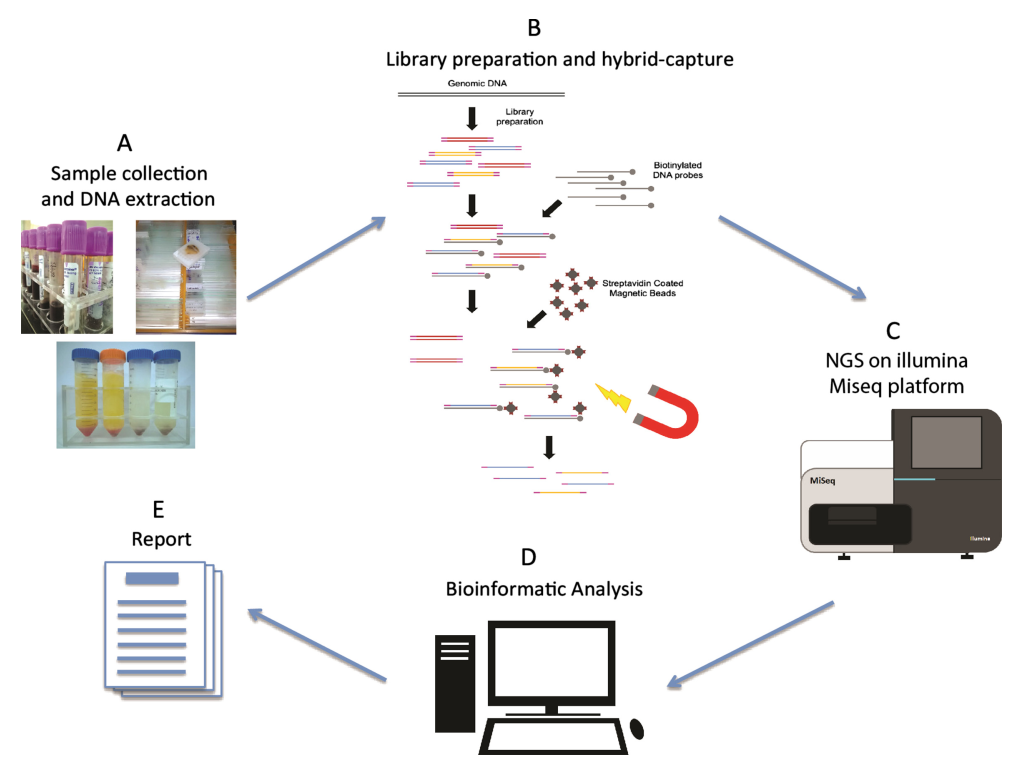
Figure 1. Next-generation sequencing workflow[2]
1. DNA library construction and its key enzymes
2. RNA library construction and its key enzymes
3. Guideline for NGS core enzymes in DNA & RNA library construction
1. DNA library construction and its key enzymes
In the process of DNA library construction, TA clone ligation adapter library construction is the most commonly used technology means at present. The main library construction process is as follows:

Figure 2. DNA library construction process (Illumina)
1.1 DNA Fragmentation
The current sequencers typically have a sequencing length in the range of 150-500 base pairs (bp). As a result, it becomes necessary to employ mechanical or enzymatic fragmentation methods to break down large genomic DNA fragments into smaller ones. Mechanical fragmentation can lead to relatively high sample loss and involves a more intricate operational process. On the other hand, enzymatic digestion is a commonly used method for fragmenting genomic DNA. In comparison to mechanical methods, enzymatic digestion is more cost-effective and straightforward, with the reaction only requiring a set period after the addition of the fragmentation enzyme.
Presently, there are primarily two types of fragments in use. One relies on the Tn5 transposase, based on transposon principles, while the other utilizes a mixture of endonucleases. However, the effectiveness of these fragments can be influenced by the GC content and base preferences of the DNA. In contrast, the fragments developed by Yeasen (Cat#12917) offer a stable digestion effect and exhibit significantly lower site preference compared to Tn5 transposase. They consistently yield excellent sequencing results for various types of DNA samples, including those from FFPE samples.
1.2 End repair, dA-Tailing
The fragmented DNA will generate 5'/3' sticky ends and blunt-end DNA, and all sticky ends need to be converted to blunt ends, including 3' overhangs removed and 5'-protruded DNA ends filled in. When using TA ligation for adapter ligation, the DNA fragment also needs to be phosphorylated at the 5' end and add "A" at the 3' end to be complementary to the adapter with the "T" sticky end. The above process is completed by the cooperation of T4 DNA polymerase, T4 polynucleotide kinase, and Taq DNA polymerase.
T4 DNA polymerase (Cat#12901) has 5'→3' DNA polymerase activity, which can catalyze the synthesis of DNA along the 5'→3' direction and fill in the 5' protruding end. At the same time, the enzyme also has 3'→5' exonuclease activity to cleave 3' overhanging ends, thereby transforming DNA fragments containing sticky ends into blunt-end DNA.
Since the 5' ends of synthetic PCR primers and adapters are usually hydroxyl groups instead of phosphate groups. Therefore, T4 polynucleotide kinase (Cat#12902) is required to catalyze the transfer of ATP γ -phosphate groups to the 5' -hydroxyl end of the oligonucleotide chain in the presence of ATP, in preparation for the next step of adapter ligation.
S-Taq DNA polymerase (Cat#13486) has 5'→3' polymerase activity, which can synthesize DNA from 5'→3' direction. Meanwhile, it has deoxynucleotidyl transferase activity, which can add a nucleotide "A" to the 3' end of the PCR product.

Figure 3. Multiple enzymes are involved in the end repair process

Figure 4. S-taq has a very high efficiency of adding A to the four bases of ATCG of the 3' end of the gene segments detected by capillary electrophoresis.
1.3 Adapter ligation
Adapters constitute a crucial component of the library. Within the realm of Illumina sequencing, the commonly employed Y-type adapters encompass P5/P7, Index, and Rd1/Rd2 SP sequences. Among these, the P5/P7 sequence serves the purpose of pairing with the sequence present on the sequencing chip, thereby anchoring the fragments to be analyzed onto the flow cell to execute bridge amplification. The Index sequence is utilized for distinguishing between different samples within the mixed library subjected to sequencing, while Rd1/Rd2 SP denote the regions for binding the Read1 and Read2 sequencing primers.
For the task of adapter ligation, T4 DNA ligase (Cat#12996) is the standard choice. It exhibits the capability to repair single-stranded nicks found in double-stranded DNA and reconnect adjacent nucleotides.
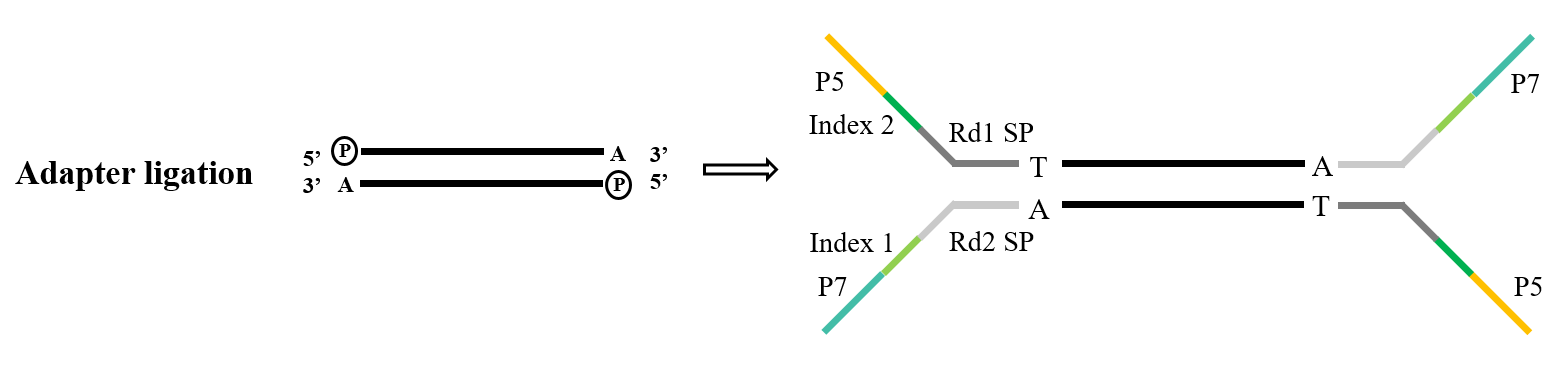
Figure 5. General adapter ligation process(Illumina)

Figure 6. Verification of T4 DNA Ligase Mutants by Ligating 170-bp DNA with 80-bp Adapters.
1.4 PCR amplification
Obtain enough DNA sequences with adapters through PCR reaction, and complete the sequencing of the sample nucleic acid sequence on the machine. Hieff CanaceTM Pro High-Fidelity DNA Polymerase (Cat#13476) commonly used in PCR has 5'→3' polymerase activity and can synthesize DNA in the 5'→3' direction. In addition, it also has the activity of 3'→5' exonuclease, which can correct the wrong incorporation of bases during the amplification process, to amplify DNA fragments rapidly and with high fidelity.
2. RNA library construction and its key enzymes
According to the types of RNA, the construction of an RNA library can be divided into mRNA library, LncRNA library, etc. Conventional RNA library includes the following processes:
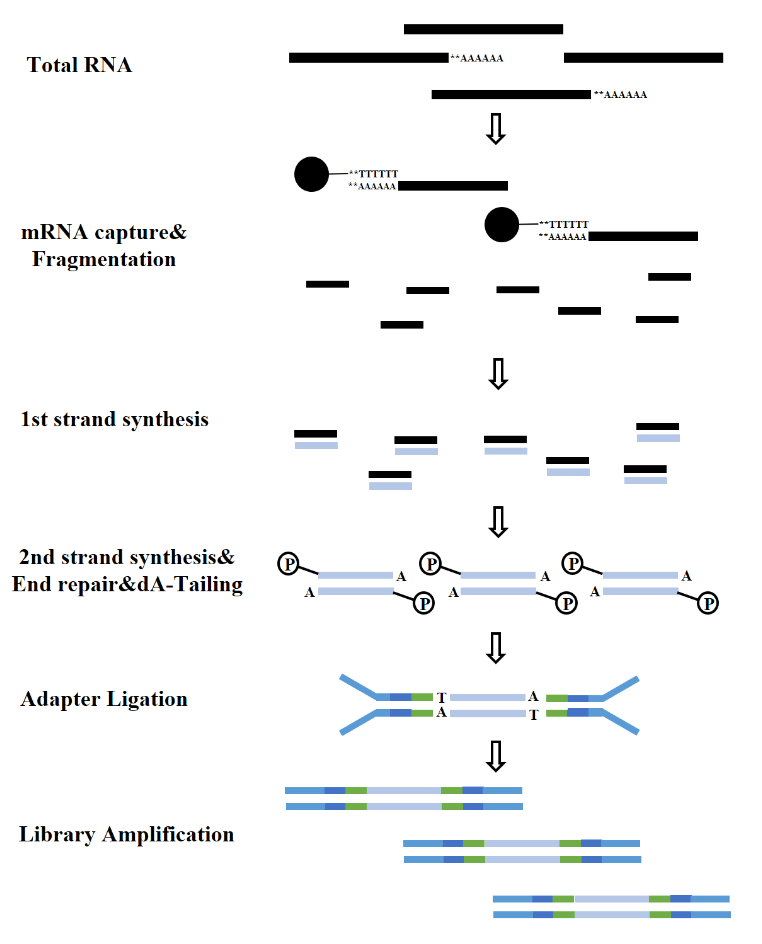
Figure 7. mRNA library construction process(Illumina)
2.1 RNA enrichment
Whether dealing with eukaryotes or prokaryotes, ribosomal RNA (rRNA) stands out as the most abundant RNA, constituting up to 80% of the total RNA content. When sequencing the total RNA of a sample directly, a substantial portion of the sequencing data will be related to rRNA. To mitigate this interference, the method of RNA enrichment must be employed. There are two primary methods for this: mRNA enrichment based on oligo-dT and rRNA depletion methods.
In eukaryotes, mRNA exhibits a distinct poly(A) structure at the 3' end. Oligo-dT beads can be employed to capture all mRNA transcribed from the sample, making it suitable for transcriptional analysis, especially with high-quality RNA samples. On the other hand, rRNA depletion methods have more lenient requirements on sample quality and can be applied to both low-quality samples (e.g., FFPE samples) and high-quality RNA samples, as well as prokaryotic samples. The commonly used commercial approach involves the use of RNase H digestion to remove rRNA, following these specific steps:
- Synthesize specific oligonucleotide probes designed to bind to rRNA.
- Employ RNase H (Cat#12906), which is capable of degrading RNA in the RNA-DNA hybrid strand, to selectively remove the rRNA bound to the probes.
- Finally, digest the DNA probes with DNase I (Cat#10325), which can degrade both single- and double-stranded DNA, effectively eliminating rRNA. For more information about DNase I, you can follow this link.
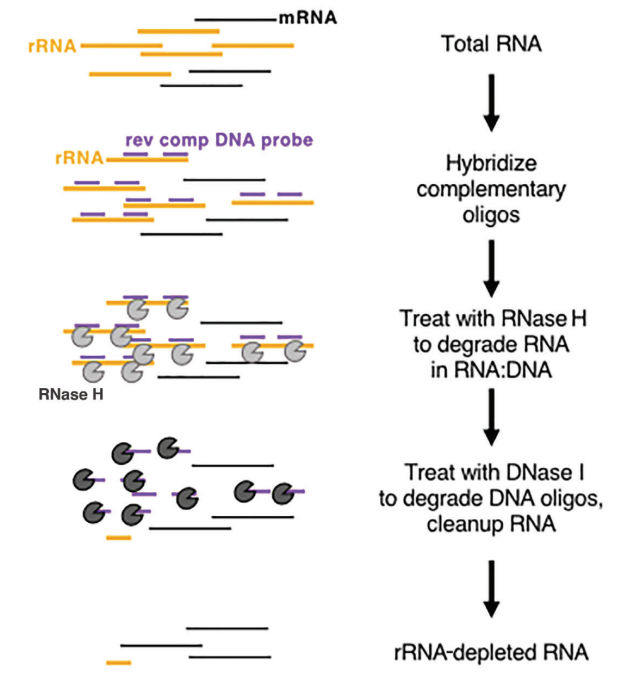
Figure 8: Schematic diagram of enzyme-based rRNA depletion[5]
2.2 RNA Fragmentation
Usually, under the action of divalent metal cations and high temperature, large fragments of RNA are broken into small fragments.
2.3 1st strand cDNA synthesis
Reverse transcription of the obtained target RNA into the first strand of cDNA. Because RNA is easily degraded by RNases present in the environment, the use of RNase Inhibitor (Cat#14672) during reverse transcription can inhibit the activity of these enzymes and protect RNA from RNase degradation. At the same time, reverse transcriptase (Cat#11112) was used to reverse transcribe the template RNA into cDNA. The reverse transcriptase has RNA-dependent DNA polymerase activity and can use RNA as a template to synthesize a cDNA in the 5'→3' direction. The single strand of DNA is complementary to the RNA template.
During the 1st strand cDNA synthesis, the incorporation of actinomycin D has undeniably improved the construction of strand-specific libraries, significantly enhancing chain specificity. This innovation has streamlined the experimental process, simplifying it for researchers.
However, actinomycin D does have its drawbacks: it exhibits toxicity and requires protection from light. In today's landscape of increasing demand for premixed and plate library constructing kits, the necessity to shield against light poses limitations on plate kit advancements.
Fortunately, Yeasen ZymeEditor platform has introduced a groundbreaking MMLV enzyme mutant (Inquiry) that replaces the function of actinomycin D. A new Kit (Cat: 12340ES) has been developed with odorless, non-toxic, and no need to avoid light. It offers superior chain specificity, eliminating concerns related to health and light sensitivity.

Figure 9: Engineering of MMLV to identify MMLV mutants which could contribute to Standed RNA-seq
2.4 2nd strand cDNA synthesis
The single-strand cDNA produced through reverse transcription is highly unstable, necessitating the immediate synthesis of the second strand of cDNA under the influence of DNA polymerase I. During this second strand synthesis, RNase H comes into play by removing the RNA strand from the RNA-DNA hybrid structure. It works in concert with DNA polymerase I (Cat#12903) to facilitate the catalytic synthesis of the complementary second strand of cDNA. DNA polymerase I possesses 5'→3' DNA polymerase activity and, guided by a template and primer, synthesizes a sequence that complements the single-strand cDNA in the 5'→3' direction.
The subsequent steps in the process include end repair, dA-Tailing, adapter ligation, and PCR amplification, all of which are detailed in the DNA library construction procedure and need not be reiterated here. It's worth noting that once reverse transcription is completed, there's no need for further fragmentation of the nucleic acid fragment.
3. Guideline for NGS core enzymes in DNA & RNA library construction
Yeasen is a biotechnology company engaged in the research, development, production, and sales of three major biological reagents: molecules, proteins, and cells. Yeasen Biotech company produces a variety of enzymes related to NGS library construction. You can choose the most suitable library construction product from the chart below.
Table 1. Guideline for NGS core enzymes in DNA & RNA library construction
|
Type |
Product positioning |
Product name |
Cat# |
|
RNA library construction |
rRNA depletion/2nd strand cDNA synthesis |
12906ES |
|
|
rRNA depletion |
10325ES |
||
|
1st strand cDNA synthesis |
14672ES |
||
|
11112ES |
|||
|
2nd strand cDNA synthesis |
12903ES |
||
|
RNA library construction & DNA library construction |
End repair |
12901ES |
|
|
12902ES |
|||
|
dA-Tailing |
13486ES |
||
|
Adapter ligation |
10301ES |
||
|
PCR amplification |
2×Super Canace® II High-Fidelity Mix for Library Amplification |
12621ES |
Table2. DNA & RNA Library Prep Kit
| Name | Cat# | Notes | |
| DNA | Hieff NGS DNA Library Prep Kit | 13577ES | Tumor/ Mechanic method |
| Hieff NGS OnePot Pro DNA Library Prep Kit V2 | 12194ES | Tumor/ Enzymetic method | |
| Hieff NGS OnePot II DNA Library Prep Kit for Illumina | 13490ES | Pathgen/ Enzymetic/ regular time (140min) | |
| Hieff NGS OnePot Flash DNA Library Prep Kit | 12316ES | Pathgen/ Enzymetic/ Ultrafast (100min) | |
| Hieff NGS DNA&RNA Library Co-Prep Kit V2 | 12305ES | Pathgen/ Enzymetic/ DNA & RNA Co-Prep | |
| RNA | Hieff NGS Ultima Dual-mode mRNA Library Prep Kit | 12308ES | Without oligo dT magnetic beads, 11 tubes |
| Hieff NGS Ultima Dual-mode mRNA Library Prep Kit | 12309ES | oligo dT magnetic beads plus, 14 tubes | |
| Hieff NGS® Ultima Dual-mode RNA Library Prep Kit | 12310ES | Premixed version, 5 tubes | |
| Hieff NGS ® EvoMax RNA Library Prep Kit(Premixed version)(actinomycin D Free) | 12340ES | Premixed version, (Actinomycin D Free) | |
| Hieff NGS® MaxUp rRNA Depletion Kit (Plant) | 12254ES | Plant | |
| Hieff NGS® MaxUp Human rRNA Depletion Kit (rRNA & ITS/ETS) | 12257ES | Human |
References:
[1] Mardis, Elaine R. Next-Generation Sequencing Platforms[J]. Annual Review of Analytical Chemistry, 2013, 6(1):287-303.
[2] Gulilat M, Lamb T, Teft W A, et al. Targeted next generation sequencing as a tool for recision medicine[J]. BMC Medical Genomics, 2019, 12(1):81.
[3] Lundberg K S, Dan D S, Adams M, et al. High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus[J]. Gene, 1991, 108(1):1-6.
[4] Miyazaki K. Random DNA fragmentation with endonuclease V: application to DNA shuffling[J]. Nucleic Acids Research, 2002, 30(24):e139.
[5] Baldwin A, Morris A R, Mukherjee N. An Easy, Cost-Effective, and Scalable Method to Deplete Human Ribosomal RNA for RNA-seq[J]. Current Protocols, 2021, 1(6):e176.