Il sequenziamento ad alta produttività, spesso definito tecnologia di sequenziamento di nuova generazione (NGS), rappresenta un significativo balzo in avanti rispetto ai metodi iniziali di sequenziamento del DNA, come il sequenziamento Sanger. L'NGS consente la profilazione simultanea di centinaia di migliaia, se non milioni, di sequenze di molecole di acido nucleico. I suoi meriti includono una produttività eccezionale, un rapporto costi-efficacia, una scalabilità e un ampio spettro di applicazioni, che lo rendono la tecnologia di sequenziamento predominante in tutto il mondo.
Il flusso di lavoro del sequenziamento NGS comprende quattro fasi principali: preparazione del campione, costruzione della libreria, sequenziamento e analisi dei dati. Al centro della costruzione della libreria c'è l'aggiunta di sequenze adattatrici standardizzate della piattaforma NGS a entrambe le estremità del DNA genomico frammentato. Questa fase mira a generare un'ampia fornitura di molecole di acido nucleico della libreria, preparate per il sequenziamento sullo strumento NGS tramite amplificazione PCR. A seconda della natura del campione, la costruzione della libreria NGS può essere categorizzata in costruzione della libreria DNA e costruzione della libreria RNA. Gli enzimi svolgono un ruolo fondamentale in questi esperimenti interconnessi. Quindi, quali enzimi chiave sono coinvolti nel processo di costruzione della libreria?
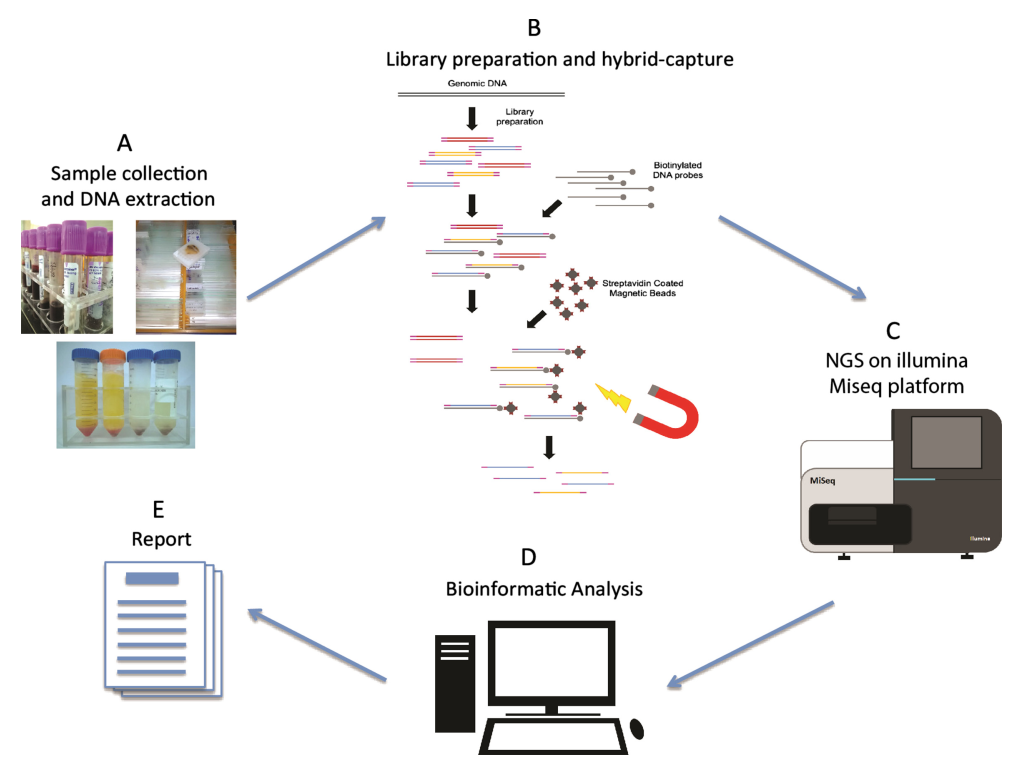
Figura 1. Flusso di lavoro del sequenziamento di nuova generazione[2]
1. Costruzione della libreria del DNA e dei suoi enzimi chiave
2. Costruzione della libreria di RNA e dei suoi enzimi chiave
3. Linee guida per gli enzimi principali NGS nella costruzione di librerie di DNA e RNA
1. Costruzione della libreria del DNA e dei suoi enzimi chiave
Nel processo di costruzione della libreria di DNA, la costruzione della libreria di adattatori di legatura dei cloni TA è il mezzo tecnologico più comunemente utilizzato al momento. Il processo principale di costruzione della libreria è il seguente:

Figura 2. Processo di costruzione della libreria del DNA (Illumina)
1.1 Frammentazione del DNA
Gli attuali sequenziatori hanno in genere una lunghezza di sequenziamento nell'intervallo di 150-500 coppie di basi (bp). Di conseguenza, diventa necessario impiegare metodi di frammentazione meccanica o enzimatica per scomporre grandi frammenti di DNA genomico in frammenti più piccoli. La frammentazione meccanica può portare a una perdita di campione relativamente elevata e comporta un processo operativo più intricato. D'altro canto, la digestione enzimatica è un metodo comunemente utilizzato per frammentare il DNA genomico. Rispetto ai metodi meccanici, la digestione enzimatica è più conveniente e semplice, con la reazione che richiede solo un periodo di tempo stabilito dopo l'aggiunta dell'enzima di frammentazione.
Attualmente, sono principalmente due i tipi di frammenti in uso. Uno si basa sulla transposasi Tn5, basata sui principi dei trasposoni, mentre l'altro utilizza una miscela di endonucleasi. Tuttavia, l'efficacia di questi frammenti può essere influenzata dal contenuto di GC e dalle preferenze di base del DNA. Al contrario, i frammenti sviluppati da Yeasen (Cat#12917) offrono un effetto di digestione stabile e mostrano una preferenza di sito significativamente inferiore rispetto alla transposasi Tn5. Forniscono costantemente risultati di sequenziamento eccellenti per vari tipi di campioni di DNA, inclusi quelli da campioni FFPE.
1.2 Riparazione finale, dA-Tailing
Il DNA frammentato genererà estremità appiccicose 5'/3' e DNA con estremità smussata, e tutte le estremità appiccicose devono essere convertite in estremità smussate, comprese le sporgenze 3' rimosse e le estremità del DNA sporgenti 5' riempite. Quando si utilizza la legatura TA per la legatura dell'adattatore, il frammento di DNA deve anche essere fosforilato all'estremità 5' e aggiungere "A" all'estremità 3' per essere complementare all'adattatore con l'estremità appiccicosa "T".Il processo sopra descritto è completato dalla cooperazione della DNA polimerasi T4, della polinucleotide chinasi T4 e Tacca DNA polimerasi.
T4 DNA polimerasi (Cat#12901) ha attività di DNA polimerasi 5'→3', che può catalizzare la sintesi del DNA lungo la direzione 5'→3' e riempire l'estremità sporgente 5'. Allo stesso tempo, l'enzima ha anche attività esonucleasica 3'→5' per scindere le estremità sporgenti 3', trasformando così i frammenti di DNA contenenti estremità appiccicose in DNA a estremità smussata.
Poiché le estremità 5' dei primer e degli adattatori PCR sintetici sono solitamente gruppi idrossilici anziché gruppi fosfato. Pertanto, la polinucleotide chinasi T4 (Cat#12902) è necessaria per catalizzare il trasferimento dei gruppi γ-fosfato ATP all'estremità 5'-idrossilica della catena oligonucleotidica in presenza di ATP, in preparazione per il passaggio successivo della legatura dell'adattatore.
S-Taq DNA polimerasi (Cat#13486) ha attività polimerasica 5'→3', che può sintetizzare il DNA dalla direzione 5'→3'. Nel frattempo, ha attività deossinucleotidil transferasi, che può aggiungere un nucleotide "A" all'estremità 3' del prodotto PCR.

Figura 3. Nel processo di riparazione finale sono coinvolti molteplici enzimi

Figura 4. S-taq ha un'efficienza molto elevata nell'aggiungere A alle quattro basi dell'ATCG dell'estremità 3' dei segmenti genici rilevati mediante elettroforesi capillare.
1.3 Legatura dell'adattatore
Gli adattatori costituiscono una componente cruciale della libreria. Nell'ambito del sequenziamento Illumina, gli adattatori di tipo Y comunemente impiegati comprendono le sequenze P5/P7, Index e Rd1/Rd2 SP. Tra queste, la sequenza P5/P7 serve allo scopo di associarsi alla sequenza presente sul chip di sequenziamento, ancorando così i frammenti da analizzare sulla cella di flusso per eseguire l'amplificazione a ponte. La sequenza Index viene utilizzata per distinguere tra diversi campioni all'interno della libreria mista sottoposta a sequenziamento, mentre Rd1/Rd2 SP denotano le regioni per il legame dei primer di sequenziamento Read1 e Read2.
Per il compito di legatura dell'adattatore, Ligasi del DNA T4 (Cat#12996) è la scelta standard. Presenta la capacità di riparare i tagli a singolo filamento presenti nel DNA a doppio filamento e di ricollegare i nucleotidi adiacenti.
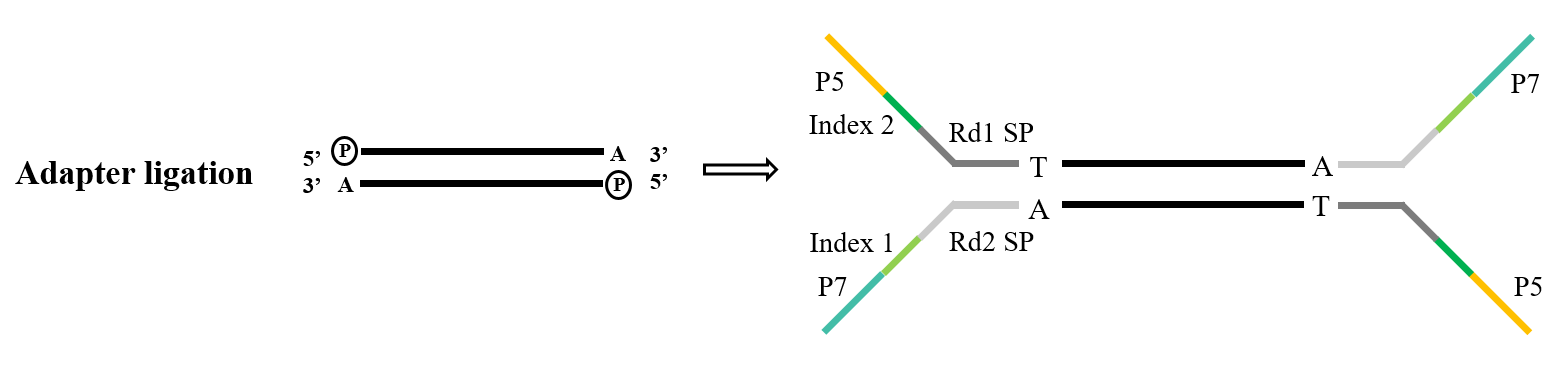
Figura 5. Processo generale di legatura dell'adattatore (Illumina)

Figura 6. Verifica dei mutanti della DNA ligasi T4 mediante la legatura di DNA da 170 bp con adattatori da 80 bp.
1.4 Amplificazione PCR
Ottieni sufficienti sequenze di DNA con adattatori tramite reazione PCR e completa il sequenziamento della sequenza di acidi nucleici campione sulla macchina. Hieff CanaceTM La Pro High-Fidelity DNA Polymerase (Cat#13476) comunemente usata nella PCR ha attività polimerasica 5'→3' e può sintetizzare il DNA nella direzione 5'→3'. Inoltre, ha anche l'attività di esonucleasi 3'→5', che può correggere l'incorporazione errata di basi durante il processo di amplificazione, per amplificare rapidamente e con alta fedeltà i frammenti di DNA.
2. Costruzione della libreria di RNA e dei suoi enzimi chiave
A seconda dei tipi di RNA, la costruzione di una libreria di RNA può essere suddivisa in libreria di mRNA, libreria di LncRNA, ecc. La libreria di RNA convenzionale include i seguenti processi:
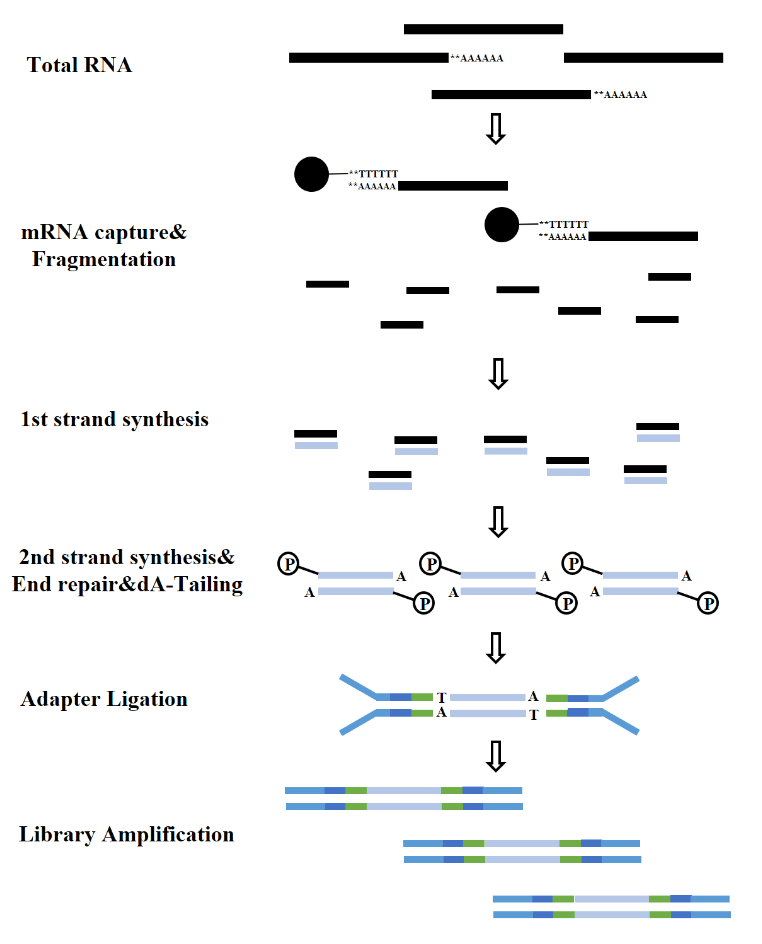
Figura 7. Processo di costruzione della libreria mRNA (Illumina)
2.1 Arricchimento dell'RNA
Che si tratti di eucarioti o procarioti, l'RNA ribosomiale (rRNA) si distingue come l'RNA più abbondante, costituendo fino all'80% del contenuto totale di RNA. Quando si sequenzia direttamente l'RNA totale di un campione, una parte sostanziale dei dati di sequenziamento sarà correlata all'rRNA. Per mitigare questa interferenza, deve essere impiegato il metodo di arricchimento dell'RNA. Esistono due metodi principali per questo: arricchimento dell'mRNA basato su oligo-dT e metodi di deplezione dell'rRNA.
Negli eucarioti, l'mRNA presenta una distinta struttura poli(A) all'estremità 3'. Le perle Oligo-dT possono essere impiegate per catturare tutto l'mRNA trascritto dal campione, rendendolo adatto per l'analisi trascrizionale, specialmente con campioni di RNA di alta qualità. D'altro canto, i metodi di deplezione dell'rRNA hanno requisiti più indulgenti sulla qualità del campione e possono essere applicati sia a campioni di bassa qualità (ad esempio, campioni FFPE) sia a campioni di RNA di alta qualità, così come a campioni procariotici. L'approccio commerciale comunemente utilizzato prevede l'uso della digestione con RNasi H per rimuovere l'rRNA, seguendo questi passaggi specifici:
- Sintetizzare sonde oligonucleotidiche specifiche progettate per legarsi all'rRNA.
- Utilizzare RNasi H (Cat#12906), in grado di degradare l'RNA nel filamento ibrido RNA-DNA, per rimuovere selettivamente l'rRNA legato alle sonde.
- Infine, digerire le sonde del DNA con DNasi I (Cat#10325), che può degradare sia il DNA a singolo che a doppio filamento, eliminando efficacemente l'rRNA. Per maggiori informazioni sulla DNasi I, puoi seguire questo link.
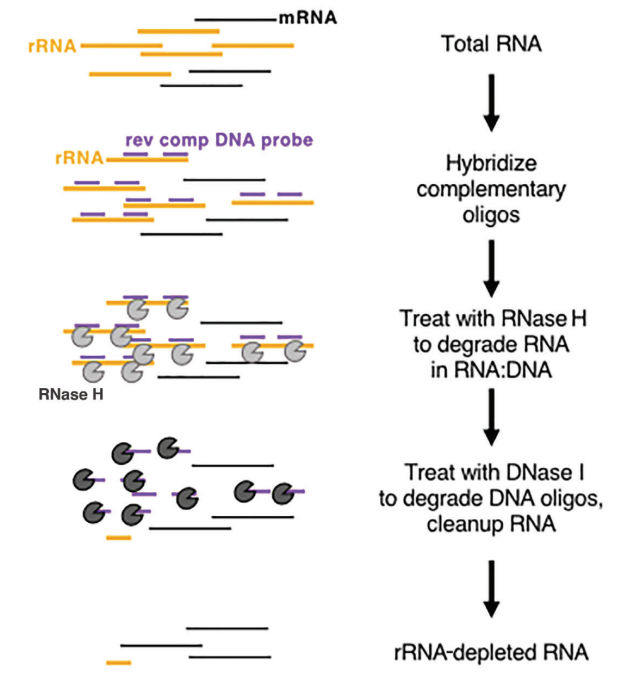
Figura 8: Diagramma schematico della deplezione dell'rRNA basata sugli enzimi[5]
2.2 Frammentazione dell'RNA
Solitamente, sotto l'azione di cationi metallici bivalenti e di alte temperature, grandi frammenti di RNA vengono scomposti in frammenti più piccoli.
2.3 Sintesi del cDNA del 1° filamento
Trascrizione inversa dell'RNA bersaglio ottenuto nel primo filamento di cDNA. Poiché l'RNA viene facilmente degradato dalle RNasi presenti nell'ambiente, l'uso di Inibitore RNasi (Cat#14672) durante la trascrizione inversa può inibire l'attività di questi enzimi e proteggere l'RNA dalla degradazione della RNasi. Allo stesso tempo, trascrittasi inversa (Cat#11112) è stato utilizzato per trascrivere inversamente l'RNA stampo in cDNA. La trascrittasi inversa ha attività di DNA polimerasi RNA-dipendente e può usare l'RNA come stampo per sintetizzare un cDNA nella direzione 5'→3'. Il singolo filamento di DNA è complementare allo stampo di RNA.
Durante il 1° sintesi di cDNA a filamento, l'incorporazione di actinomicina D ha innegabilmente migliorato la costruzione di librerie specifiche per filamento, potenziando significativamente la specificità della catena. Questa innovazione ha semplificato il processo sperimentale, semplificandolo per i ricercatori.
Tuttavia, l'actinomicina D ha i suoi svantaggi: è tossica e richiede protezione dalla luce. Nel panorama odierno di crescente domanda di kit di costruzione di librerie premiscelate e su piastra, la necessità di schermare dalla luce pone delle limitazioni ai progressi dei kit su piastra.
Fortunatamente, la piattaforma Yeasen ZymeEditor ha introdotto un rivoluzionario mutante enzimatico MMLV (Inquiry) che sostituisce la funzione dell'actinomicina D. Un nuovo Il kit (Cat: 12340ES) è stato sviluppato con materiali inodori, non tossici e no bisogno di evitare luce. Offre una specificità di catena superiore, eliminando le preoccupazioni relative alla salute e alla sensibilità alla luce.

Figura 9: Ingegneria di MMLV per identificare i mutanti MMLV che potrebbero contribuire a Standed RNA-seq
2.4 Sintesi del cDNA del 2° filamento
Il cDNA a singolo filamento prodotto tramite trascrizione inversa è altamente instabile, rendendo necessaria la sintesi immediata del secondo filamento di cDNA sotto l'influenza della DNA polimerasi I. Durante questa sintesi del secondo filamento, la RNasi H entra in gioco rimuovendo il filamento di RNA dalla struttura ibrida RNA-DNA. Lavora di concerto con DNA polimerasi I (Cat#12903) per facilitare la sintesi catalitica del secondo filamento complementare di cDNA. La DNA polimerasi I possiede attività di DNA polimerasi 5'→3' e, guidata da uno stampo e da un primer, sintetizza una sequenza che completa il cDNA a singolo filamento nella direzione 5'→3'.
I passaggi successivi del processo includono la riparazione delle estremità, il dA-Tailing, la legatura dell'adattatore e l'amplificazione tramite PCR, tutti dettagliati nella procedura di costruzione della libreria di DNA e non è necessario ripeterli qui. Vale la pena notare che una volta completata la trascrizione inversa, non c'è bisogno di un'ulteriore frammentazione del frammento di acido nucleico.
3. Linee guida per gli enzimi principali NGS nella costruzione di librerie di DNA e RNA
Yeasen è un'azienda biotecnologica impegnata nella ricerca, sviluppo, produzione e vendita di tre principali reagenti biologici: molecole, proteine e cellule. L'azienda Yeasen Biotech produce una varietà di enzimi correlati alla costruzione di librerie NGS. Puoi scegliere il prodotto di costruzione di librerie più adatto dalla tabella sottostante.
Tabella 1.Linee guida per gli enzimi principali NGS nella costruzione di librerie di DNA e RNA
| Tipo | Posizionamento del prodotto | Nome del prodotto | Gatto# |
| Libreria di RNA costruzione | RNA ricostituito deplezione/sintesi del cDNA del 2° filamento | 12906ES | |
| RNA ricostituito esaurimento | 10325ES | ||
| Sintesi del cDNA del 1° filamento | 14672ES | ||
| 11112ES | |||
| Sintesi del cDNA del 2° filamento | 12903ES | ||
| Libreria di RNA costruzione & DNA biblioteca costruzione | Riparazione finale | 12901ES | |
| 12902ES | |||
| dA-Coda | 13486ES | ||
| Legatura dell'adattatore | 10301ES | ||
| PCR amplificazione | 2×Super Canace® II High-Fidelity Mix per l'amplificazione della libreria | 12621ES |
Tabella 2.Libreria DNA e RNA Kit di preparazione
| Nome | Gatto# | Appunti | |
| Il DNA | Kit di preparazione della libreria DNA NGS Hieff | 13577ES | Metodo Tumore/Meccanico |
| Kit di preparazione della libreria DNA Hieff NGS OnePot Pro V2 | 12194ES | Metodo tumorale/enzimatico | |
| Hieff NGS OnePot II Kit di preparazione della libreria del DNA per Illumina | 13490ES | Patogeno/enzimatico/tempo regolare (140 min) | |
| Kit di preparazione della libreria DNA Flash OnePot NGS Hieff | 12316ES | Patogeno/Enzimatico/Ultraveloce (100 minuti) | |
| Kit di co-preparazione della libreria Hieff NGS DNA&RNA V2 | 12305ES | Co-preparazione di patogeni/enzimatici/DNA e RNA | |
| RNA | Kit di preparazione della libreria mRNA a doppia modalità Hieff NGS Ultima | 12308ES | Senza sfere magnetiche oligo dT, 11 tubi |
| Kit di preparazione della libreria mRNA a doppia modalità Hieff NGS Ultima | 12309ES | perle magnetiche oligo dT plus, 14 tubi | |
| Kit di preparazione della libreria RNA a doppia modalità Hieff NGS® Ultima | 12310ES | Versione premiscelata, 5 tubi | |
| Kit di preparazione della libreria RNA Hieff NGS ® EvoMax (versione premiscelata) (actinomicina D Gratuito) | 12340ES | Versione premiscelata, (Actinomicina D Gratuito) | |
| Kit di deplezione dell'rRNA Hieff NGS® MaxUp (pianta) | 12254ES | Pianta | |
| Kit di deplezione dell'rRNA umano Hieff NGS® MaxUp (rRNA e ITS/ETS) | 12257ES | Umano |
Riferimenti:
[1] Mardis, Elaine R. Piattaforme di sequenziamento di nuova generazione[J]. Revisione annuale di chimica analitica, 2013, 6(1):287-303.
[2] Gulilat M, Lamb T, Teft WA, et al. Sequenziamento mirato di nuova generazione come strumento per la medicina di precisione[J]. BMC Medical Genomics, 2019, 12(1):81.
[3] Lundberg KS, Dan DS, Adams M, et al. Amplificazione ad alta fedeltà utilizzando una DNA polimerasi termostabile isolata da Pyrococcus furiosus[J]. Gene, 1991, 108(1):1-6.
[4] Miyazaki K. Frammentazione casuale del DNA con endonucleasi V: applicazione allo shuffling del DNA[J]. Nucleic Acids Research, 2002, 30(24):e139.
[5] Baldwin A, Morris AR, Mukherjee N. Un metodo semplice, conveniente e scalabile per esaurire l'RNA ribosomiale umano per RNA-seq[J]. Current Protocols, 2021, 1(6):e176.