La secuenciación de alto rendimiento, a menudo denominada tecnología de secuenciación de próxima generación (NGS), representa un avance significativo con respecto a los métodos iniciales de secuenciación de ADN, como la secuenciación de Sanger. La NGS permite la elaboración simultánea de perfiles de cientos de miles, si no millones, de secuencias de moléculas de ácidos nucleicos. Entre sus ventajas se incluyen un rendimiento excepcional, rentabilidad, escalabilidad y un amplio espectro de aplicaciones, lo que la ha convertido en la tecnología de secuenciación predominante en todo el mundo.
El flujo de trabajo de secuenciación NGS comprende cuatro fases principales: preparación de la muestra, construcción de la biblioteca, secuenciación y análisis de datos. Un aspecto central de la construcción de la biblioteca es la unión de secuencias adaptadoras de la plataforma NGS estandarizadas a ambos extremos del ADN genómico fragmentado. Este paso tiene como objetivo generar un amplio suministro de moléculas de ácidos nucleicos de la biblioteca, preparadas para la secuenciación en el instrumento NGS mediante amplificación por PCR. Según la naturaleza de la muestra, la construcción de la biblioteca NGS se puede clasificar en construcción de biblioteca de ADN y construcción de biblioteca de ARN. Las enzimas desempeñan un papel fundamental en estos experimentos interconectados. Entonces, ¿qué enzimas clave participan en el proceso de construcción de la biblioteca?
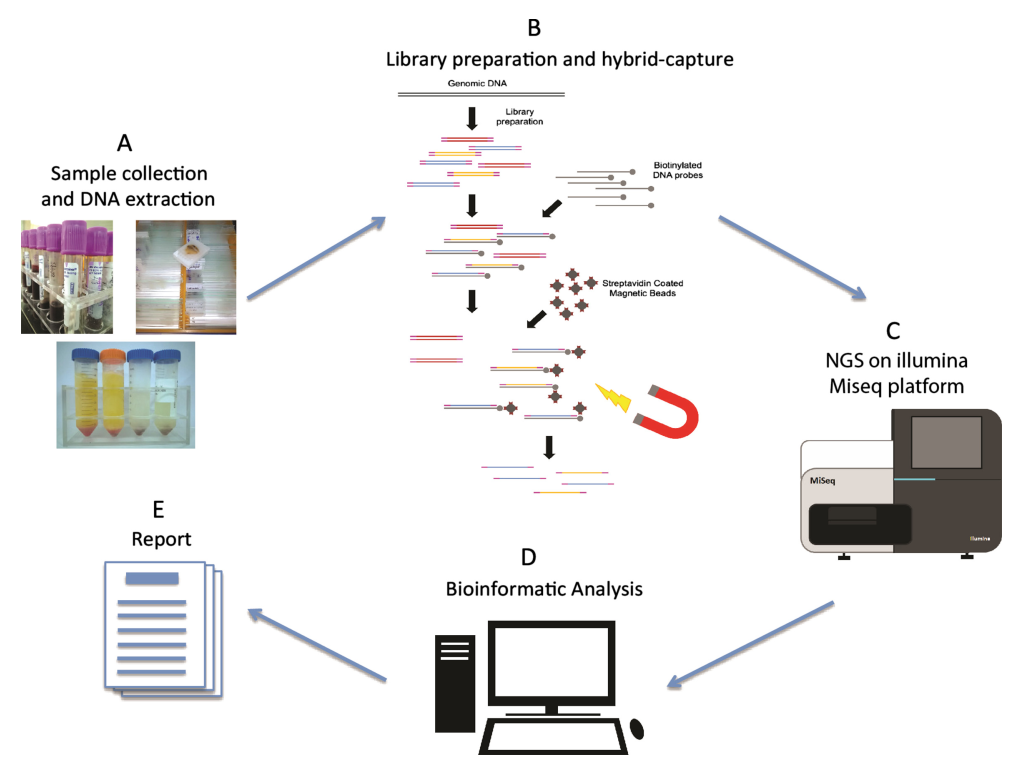
Figura 1. Flujo de trabajo de secuenciación de próxima generación[2]
1. Construcción de bibliotecas de ADN y sus enzimas clave
2. Construcción de bibliotecas de ARN y sus enzimas clave
3. Directrices para las enzimas centrales de NGS en la construcción de bibliotecas de ADN y ARN
1. Construcción de bibliotecas de ADN y sus enzimas clave
En el proceso de construcción de bibliotecas de ADN, la construcción de bibliotecas de adaptadores de ligación de clones TA es el medio tecnológico más utilizado en la actualidad. El proceso principal de construcción de bibliotecas es el siguiente:

Figura 2. Proceso de construcción de la biblioteca de ADN (Illumina)
1.1 Fragmentación del ADN
Los secuenciadores actuales suelen tener una longitud de secuenciación de entre 150 y 500 pares de bases (pb). Como resultado, se hace necesario emplear métodos de fragmentación mecánica o enzimática para descomponer fragmentos grandes de ADN genómico en fragmentos más pequeños. La fragmentación mecánica puede provocar una pérdida de muestra relativamente alta e implica un proceso operativo más complejo. Por otro lado, la digestión enzimática es un método de uso común para fragmentar el ADN genómico. En comparación con los métodos mecánicos, la digestión enzimática es más rentable y sencilla, ya que la reacción solo requiere un período determinado después de la adición de la enzima de fragmentación.
En la actualidad, se utilizan principalmente dos tipos de fragmentos. Uno se basa en la transposasa Tn5, basada en principios de transposón, mientras que el otro utiliza una mezcla de endonucleasas. Sin embargo, la eficacia de estos fragmentos puede verse influida por el contenido de GC y las preferencias de bases del ADN. Por el contrario, los fragmentos desarrollados por Yeasen (Cat#12917) ofrecen un efecto de digestión estable y muestran una preferencia de sitio significativamente menor en comparación con la transposasa Tn5. Producen constantemente excelentes resultados de secuenciación para varios tipos de muestras de ADN, incluidas las de muestras FFPE.
1.2 Reparación de extremos, dA-Tailing
El ADN fragmentado generará extremos pegajosos 5'/3' y ADN con extremos romos, y todos los extremos pegajosos deben convertirse en extremos romos, incluidos los salientes 3' eliminados y los extremos de ADN que sobresalen 5' rellenados. Cuando se utiliza la ligadura TA para la ligadura del adaptador, el fragmento de ADN también debe fosforilarse en el extremo 5' y agregar "A" en el extremo 3' para que sea complementario al adaptador con el extremo pegajoso "T".El proceso anterior se completa mediante la cooperación de la ADN polimerasa T4, la polinucleótido quinasa T4 y Taq ADN polimerasa.
ADN polimerasa T4 (Cat. n.° 12901) Tiene actividad de polimerasa de ADN 5'→3', que puede catalizar la síntesis de ADN a lo largo de la dirección 5'→3' y rellenar el extremo saliente 5'. Al mismo tiempo, la enzima también tiene actividad de exonucleasa 3'→5' para escindir los extremos salientes 3', transformando así los fragmentos de ADN que contienen extremos pegajosos en ADN de extremos romos.
Dado que los extremos 5' de los adaptadores y cebadores sintéticos para PCR suelen ser grupos hidroxilo en lugar de grupos fosfato, se requiere la polinucleótido quinasa T4 (Cat#12902) para catalizar la transferencia de grupos γ-fosfato de ATP al extremo 5'-hidroxilo de la cadena de oligonucleótidos en presencia de ATP, en preparación para el siguiente paso de la ligación del adaptador.
S-Taq ADN polimerasa (Cat#13486) tiene actividad polimerasa 5'→3', que puede sintetizar ADN en la dirección 5'→3'. Mientras tanto, tiene actividad desoxinucleotidil transferasa, que puede agregar un nucleótido "A" al extremo 3' del producto de PCR.

Figura 3. En el proceso de reparación final intervienen múltiples enzimas.

Figura 4. S-taq tiene una eficiencia muy alta en la adición de A a las cuatro bases de ATCG del extremo 3' de los segmentos genéticos detectados por electroforesis capilar.
1.3 Ligadura del adaptador
Los adaptadores constituyen un componente crucial de la biblioteca. En el ámbito de la secuenciación de Illumina, los adaptadores de tipo Y que se emplean habitualmente incluyen las secuencias P5/P7, Index y Rd1/Rd2 SP. Entre ellas, la secuencia P5/P7 sirve para emparejarse con la secuencia presente en el chip de secuenciación, anclando así los fragmentos que se van a analizar en la celda de flujo para ejecutar la amplificación en puente. La secuencia Index se utiliza para distinguir entre diferentes muestras dentro de la biblioteca mixta sujeta a secuenciación, mientras que Rd1/Rd2 SP denota las regiones para la unión de los cebadores de secuenciación Read1 y Read2.
Para la tarea de ligadura del adaptador, Ligasa de ADN T4 (Cat. n.° 12996) Es la opción estándar. Presenta la capacidad de reparar cortes monocatenarios encontrados en ADN bicatenario y reconectar nucleótidos adyacentes.
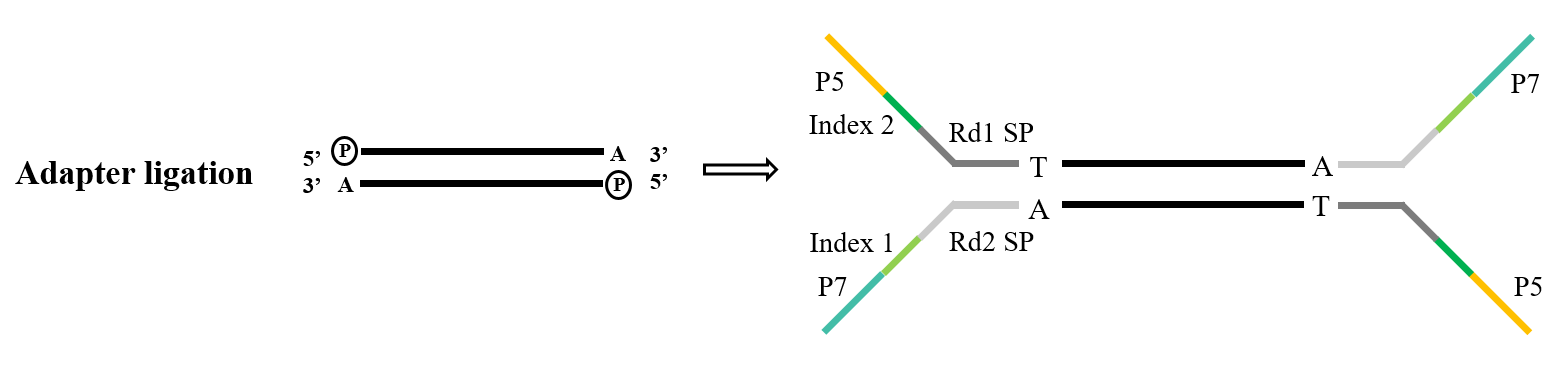
Figura 5. Proceso general de ligadura del adaptador (Illumina)

Figura 6. Verificación de mutantes de la ADN ligasa T4 mediante la ligación de ADN de 170 pb con adaptadores de 80 pb.
1.4 Amplificación por PCR
Obtenga suficientes secuencias de ADN con adaptadores mediante la reacción de PCR y complete la secuenciación de la secuencia de ácido nucleico de muestra en la máquina. Hieff CanaceMT. La ADN polimerasa Pro High-Fidelity (Cat#13476), que se utiliza habitualmente en PCR, tiene actividad polimerasa 5'→3' y puede sintetizar ADN en la dirección 5'→3'. Además, también tiene la actividad de exonucleasa 3'→5', que puede corregir la incorporación incorrecta de bases durante el proceso de amplificación, para amplificar fragmentos de ADN rápidamente y con alta fidelidad.
2. Construcción de bibliotecas de ARN y sus enzimas clave
Según los tipos de ARN, la construcción de una biblioteca de ARN se puede dividir en biblioteca de ARNm, biblioteca de LncRNA, etc. La biblioteca de ARN convencional incluye los siguientes procesos:
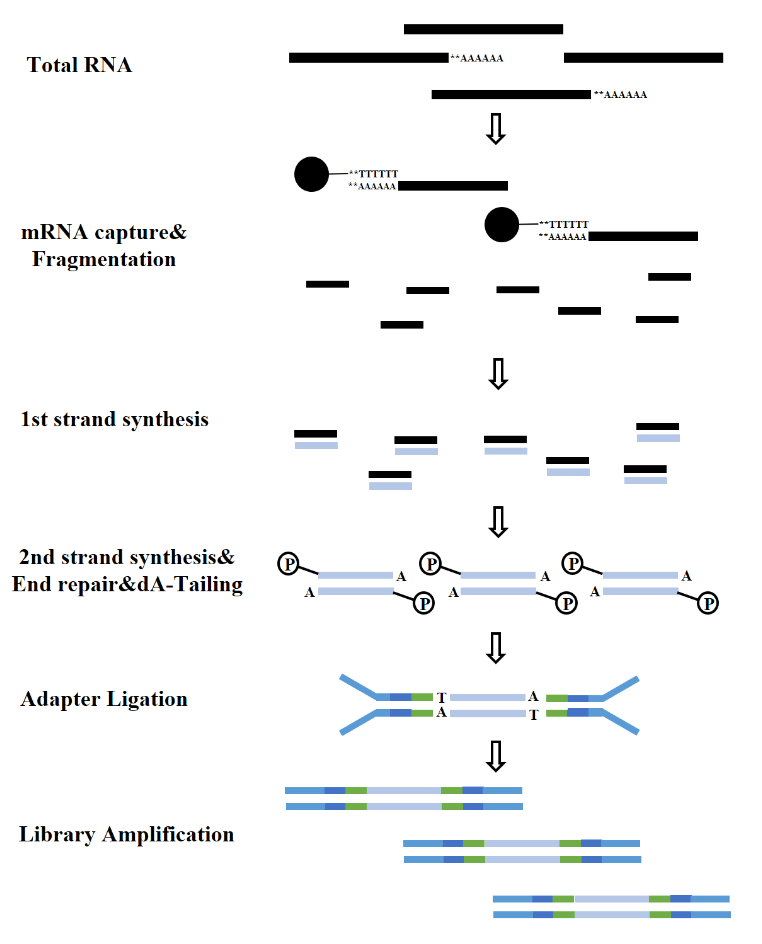
Figura 7. Proceso de construcción de la biblioteca de ARNm (Illumina)
2.1 Enriquecimiento de ARN
Ya se trate de eucariotas o procariotas, el ARN ribosómico (ARNr) se destaca como el ARN más abundante, constituyendo hasta el 80% del contenido total de ARN. Al secuenciar el ARN total de una muestra directamente, una parte sustancial de los datos de secuenciación estarán relacionados con el ARNr. Para mitigar esta interferencia, se debe emplear el método de enriquecimiento del ARN. Existen dos métodos principales para esto: el enriquecimiento del ARNm basado en oligo-dT y los métodos de depleción del ARNr.
En los eucariotas, el ARNm presenta una estructura poli(A) distintiva en el extremo 3'. Las perlas de oligo-dT se pueden emplear para capturar todo el ARNm transcrito de la muestra, lo que lo hace adecuado para el análisis transcripcional, especialmente con muestras de ARN de alta calidad. Por otro lado, los métodos de depleción de ARNr tienen requisitos más indulgentes en cuanto a la calidad de la muestra y se pueden aplicar tanto a muestras de baja calidad (por ejemplo, muestras FFPE) como a muestras de ARN de alta calidad, así como a muestras procariotas. El enfoque comercial comúnmente utilizado implica el uso de la digestión con ARNasa H para eliminar el ARNr, siguiendo estos pasos específicos:
- Sintetizar sondas de oligonucleótidos específicas diseñadas para unirse al ARNr.
- Utilice la ARNasa H (Cat#12906), que es capaz de degradar el ARN en la cadena híbrida ARN-ADN, para eliminar selectivamente el ARNr unido a las sondas.
- Finalmente, digerir las sondas de ADN con DNasa I (Cat#10325), que puede degradar tanto el ADN monocatenario como el bicatenario, eliminando eficazmente el ARNr. Para obtener más información sobre la ADNasa I, puede seguir este enlace.
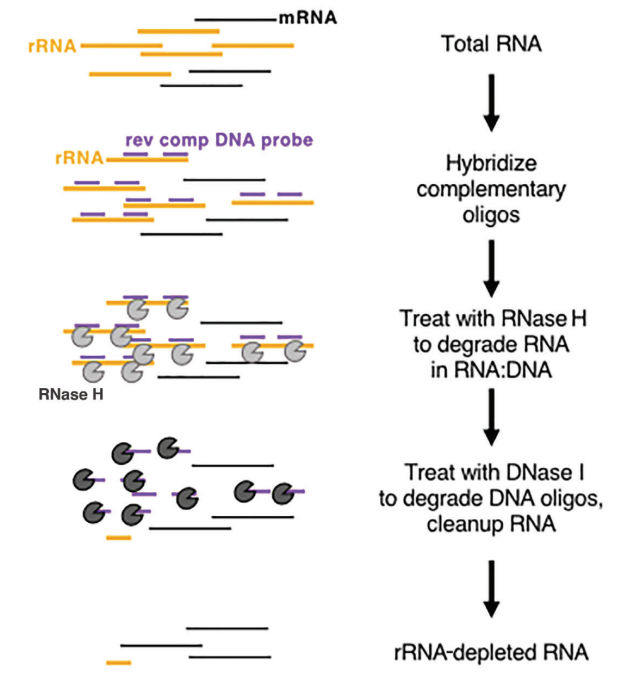
Figura 8: Diagrama esquemático de la depleción de ARNr basada en enzimas[5]
2.2 Fragmentación del ARN
Generalmente, bajo la acción de cationes metálicos divalentes y altas temperaturas, grandes fragmentos de ARN se rompen en fragmentos más pequeños.
2.3 Síntesis de ADNc de primera cadena
Transcripción inversa del ARN diana obtenido en la primera cadena de ADNc. Debido a que el ARN se degrada fácilmente por las ARNasas presentes en el medio ambiente, el uso de Inhibidor de la ARNasa (Cat. n.° 14672) Durante la transcripción inversa, pueden inhibir la actividad de estas enzimas y proteger el ARN de la degradación por ARNasa. Al mismo tiempo, transcriptasa inversa (Cat#11112) Se utilizó para transcribir de forma inversa el ARN molde en ADNc. La transcriptasa inversa tiene actividad de ADN polimerasa dependiente del ARN y puede utilizar el ARN como molde para sintetizar un ADNc en la dirección 5'→3'. La cadena simple de ADN es complementaria al molde de ARN.
Durante el 1er En la síntesis de ADNc de cadena única, la incorporación de actinomicina D ha mejorado indudablemente la construcción de bibliotecas específicas de cadena, mejorando significativamente la especificidad de la cadena. Esta innovación ha agilizado el proceso experimental, simplificándolo para los investigadores.
Sin embargo, la actinomicina D tiene sus desventajas: presenta toxicidad y requiere protección contra la luz. En el panorama actual de creciente demanda de kits de construcción de bibliotecas de placas y premezclados, la necesidad de protección contra la luz plantea limitaciones a los avances en los kits de placas.
Afortunadamente, la plataforma Yeasen ZymeEditor ha introducido un mutante revolucionario de la enzima MMLV (Investigación) que reemplaza la función de la actinomicina D. Un nuevo El kit (Cat: 12340ES) ha sido desarrollado con componentes inodoros, no tóxicos y noo necesidad de evitar Ligero. Ofrece una especificidad de cadena superior, eliminando preocupaciones relacionadas con la salud y la sensibilidad a la luz.

2.4 Síntesis de ADNc de segunda cadena
El ADNc monocatenario producido mediante transcripción inversa es altamente inestable, lo que requiere la síntesis inmediata de la segunda cadena de ADNc bajo la influencia de la ADN polimerasa I. Durante esta síntesis de la segunda cadena, la ARNasa H entra en acción eliminando la cadena de ARN de la estructura híbrida ARN-ADN. Trabaja en conjunto con ADN polimerasa I (Cat. n.° 12903) para facilitar la síntesis catalítica de la segunda cadena complementaria de ADNc. La ADN polimerasa I posee actividad de ADN polimerasa 5'→3' y, guiada por una plantilla y un cebador, sintetiza una secuencia que complementa el ADNc de cadena sencilla en la dirección 5'→3'.
Los pasos posteriores del proceso incluyen la reparación de extremos, la unión de dA, la ligadura de adaptadores y la amplificación por PCR, todos los cuales se detallan en el procedimiento de construcción de la biblioteca de ADN y no es necesario reiterarlos aquí. Vale la pena señalar que una vez que se completa la transcripción inversa, no es necesario realizar una mayor fragmentación del fragmento de ácido nucleico.
3. Directrices para las enzimas centrales de NGS en la construcción de bibliotecas de ADN y ARN
Yeasen es una empresa de biotecnología dedicada a la investigación, desarrollo, producción y venta de tres reactivos biológicos principales: moléculas, proteínas y células. La empresa Yeasen Biotech produce una variedad de enzimas relacionadas con la construcción de bibliotecas NGS. Puede elegir el producto de construcción de bibliotecas más adecuado en la siguiente tabla.
Tabla 1.Guía para las enzimas centrales de NGS en la construcción de bibliotecas de ADN y ARN
| Tipo | Posicionamiento del producto | Nombre del producto | Gato# |
| Biblioteca de ARN construcción | ARNr Agotamiento/síntesis de ADNc de segunda cadena | 12906ES | |
| ARNr agotamiento | 10325ES | ||
| Síntesis de ADNc de primera cadena | 14672ES | ||
| 11112ES | |||
| Síntesis de ADNc de segunda cadena | 12903ES | ||
| Biblioteca de ARN construcción & ADN biblioteca construcción | Finalizar la reparación | 12901ES | |
| 12902ES | |||
| dA-Cola | 13486ES | ||
| Ligadura del adaptador | 10301ES | ||
| PCR amplificación | Mezcla de alta fidelidad 2×Super Canace® II para amplificación de biblioteca | 12621ES |
Tabla 2.Biblioteca de ADN y ARN Kit de preparación
| Nombre | Gato# | Notas | |
| ADN | Kit de preparación de biblioteca de ADN NGS de Hieff | 13577ES | Método tumoral/mecánico |
| Kit de preparación de biblioteca de ADN Hieff NGS OnePot Pro V2 | 12194ES | Método enzimático/tumoral | |
| NGS OnePot de alta potencia Kit de preparación de biblioteca de ADN II para Illumina | 13490ES | Patógeno/enzimético/tiempo regular (140 min) | |
| Kit de preparación de biblioteca de ADN Hieff NGS OnePot Flash | 12316ES | Patógeno/ Enzimético/ Ultrarrápido (100 minutos) | |
| Kit de preparación conjunta para biblioteca de ADN y ARN Hieff NGS V2 | 12305ES | Preparación conjunta de ADN y ARN, enzimas y patógenos | |
| ARN | Kit de preparación de biblioteca de ARNm de modo dual Hieff NGS Ultima | 12308ES | Sin perlas magnéticas de oligo dT, 11 tubos |
| Kit de preparación de biblioteca de ARNm de modo dual Hieff NGS Ultima | 12309ES | Perlas magnéticas oligo dT plus, 14 tubos | |
| Kit de preparación de biblioteca de ARN de modo dual Hieff NGS® Ultima | 12310ES | Versión premezclada, 5 tubos | |
| Kit de preparación de biblioteca de ARN Hieff NGS ® EvoMax (versión premezclada) (actinomicina D Gratis) | 12340ES | Versión premezclada, (Actinomicina D Gratis) | |
| Kit de depleción de ARNr Hieff NGS® MaxUp (planta) | 12254ES | Planta | |
| Kit de depleción de ARNr humano Hieff NGS® MaxUp (ARNr e ITS/ETS) | 12257ES | Humano |
Referencias:
[1] Mardis, Elaine R. Plataformas de secuenciación de próxima generación[J]. Revisión anual de química analítica, 2013, 6(1):287-303.
[2] Gulilat M, Lamb T, Teft WA, et al. Secuenciación dirigida de próxima generación como herramienta para la medicina de recisión[J]. BMC Medical Genomics, 2019, 12(1):81.
[3] Lundberg KS, Dan DS, Adams M, et al. Amplificación de alta fidelidad utilizando una ADN polimerasa termoestable aislada de Pyrococcus furiosus[J]. Gene, 1991, 108(1):1-6.
[4] Miyazaki K. Fragmentación aleatoria de ADN con endonucleasa V: aplicación a la reorganización del ADN[J]. Nucleic Acids Research, 2002, 30(24):e139.
[5] Baldwin A, Morris AR, Mukherjee N. Un método fácil, rentable y escalable para agotar el ARN ribosómico humano para la secuenciación de ARN[J]. Protocolos actuales, 2021, 1(6):e176.