O sequenciamento de alto rendimento, frequentemente chamado de tecnologia de sequenciamento de próxima geração (NGS), representa um salto significativo em relação aos métodos iniciais de sequenciamento de DNA, como o sequenciamento de Sanger. O NGS permite o perfilamento simultâneo de centenas de milhares, se não milhões, de sequências de moléculas de ácido nucleico. Seus méritos incluem rendimento excepcional, custo-efetividade, escalabilidade e um amplo espectro de aplicações, estabelecendo-o como a tecnologia de sequenciamento predominante no mundo todo.
O fluxo de trabalho de sequenciamento NGS abrange quatro fases principais: preparação da amostra, construção da biblioteca, sequenciamento e análise de dados. O ponto central da construção da biblioteca é a fixação de sequências adaptadoras de plataforma NGS padronizadas em ambas as extremidades do DNA genômico fragmentado. Esta etapa visa gerar um amplo suprimento de moléculas de ácido nucleico da biblioteca, preparadas para sequenciamento no instrumento NGS por meio da amplificação por PCR. Dependendo da natureza da amostra, a construção da biblioteca NGS pode ser categorizada em construção da biblioteca de DNA e construção da biblioteca de RNA. As enzimas desempenham um papel fundamental nesses experimentos interconectados. Então, quais enzimas-chave estão envolvidas no processo de construção da biblioteca?
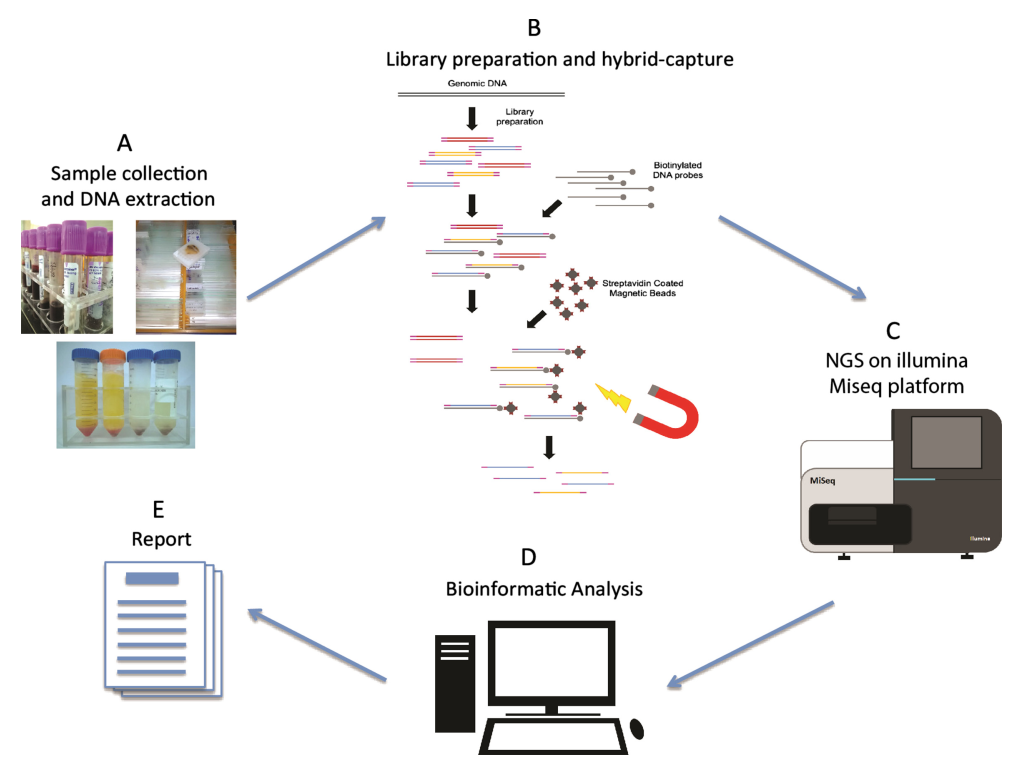
Figura 1. Fluxo de trabalho de sequenciamento de próxima geração[2]
1. Construção da biblioteca de DNA e suas principais enzimas
2. Construção da biblioteca de RNA e suas principais enzimas
3. Diretriz para enzimas centrais NGS na construção de bibliotecas de DNA e RNA
1. Construção da biblioteca de DNA e suas principais enzimas
No processo de construção de biblioteca de DNA, a construção de biblioteca de adaptador de ligação de clone TA é o meio de tecnologia mais comumente usado no momento. O principal processo de construção de biblioteca é o seguinte:

Figura 2. Processo de construção da biblioteca de DNA (Illumina)
1.1 Fragmentação de DNA
Os sequenciadores atuais normalmente têm um comprimento de sequenciamento na faixa de 150-500 pares de bases (pb). Como resultado, torna-se necessário empregar métodos de fragmentação mecânica ou enzimática para quebrar grandes fragmentos de DNA genômico em menores. A fragmentação mecânica pode levar a uma perda de amostra relativamente alta e envolve um processo operacional mais complexo. Por outro lado, a digestão enzimática é um método comumente usado para fragmentar DNA genômico. Em comparação aos métodos mecânicos, a digestão enzimática é mais econômica e direta, com a reação exigindo apenas um período definido após a adição da enzima de fragmentação.
Atualmente, há principalmente dois tipos de fragmentos em uso. Um depende da transposase Tn5, com base em princípios de transposon, enquanto o outro utiliza uma mistura de endonucleases. No entanto, a eficácia desses fragmentos pode ser influenciada pelo conteúdo de GC e preferências de base do DNA. Em contraste, os fragmentos desenvolvidos por
1.2 Reparo final, dA-Tailing
O DNA fragmentado gerará extremidades adesivas 5'/3' e DNA de extremidade romba, e todas as extremidades adesivas precisam ser convertidas em extremidades rombas, incluindo saliências 3' removidas e extremidades de DNA salientes 5' preenchidas. Ao usar a ligação TA para ligação do adaptador, o fragmento de DNA também precisa ser fosforilado na extremidade 5' e adicionar "A" na extremidade 3' para ser complementar ao adaptador com a extremidade adesiva "T".O processo acima é completado pela cooperação da T4 DNA polimerase, T4 polinucleotídeo quinase e Taq DNA polimerase.
T4 DNA polimerase (Cat#12901) tem atividade de DNA polimerase 5'→3', que pode catalisar a síntese de DNA ao longo da direção 5'→3' e preencher a extremidade saliente 5'. Ao mesmo tempo, a enzima também tem atividade de exonuclease 3'→5' para clivar extremidades salientes 3', transformando assim fragmentos de DNA contendo extremidades pegajosas em DNA de extremidade romba.
Como as extremidades 5' dos primers e adaptadores sintéticos de PCR são geralmente grupos hidroxila em vez de grupos fosfato. Portanto, a cinase polinucleotídica T4 (Cat#12902) é necessária para catalisar a transferência de grupos γ-fosfato de ATP para a extremidade 5'-hidroxila da cadeia de oligonucleotídeos na presença de ATP, em preparação para a próxima etapa da ligação do adaptador.
S-Taq DNA polimerase (Cat#13486) tem atividade de polimerase 5'→3', que pode sintetizar DNA da direção 5'→3'. Enquanto isso, tem atividade de desoxinucleotidil transferase, que pode adicionar um nucleotídeo "A" à extremidade 3' do produto de PCR.

Figura 3. Várias enzimas estão envolvidas no processo de reparo final

Figura 4. S-taq tem uma eficiência muito alta de adicionar A às quatro bases de ATCG da extremidade 3' dos segmentos gênicos detectados por eletroforese capilar.
1.3 Ligação do adaptador
Os adaptadores constituem um componente crucial da biblioteca. Dentro do reino do sequenciamento Illumina, os adaptadores do tipo Y comumente empregados abrangem sequências P5/P7, Index e Rd1/Rd2 SP. Entre elas, a sequência P5/P7 serve ao propósito de parear com a sequência presente no chip de sequenciamento, ancorando assim os fragmentos a serem analisados na célula de fluxo para executar a amplificação de ponte. A sequência Index é utilizada para distinguir entre diferentes amostras dentro da biblioteca mista submetida ao sequenciamento, enquanto Rd1/Rd2 SP denotam as regiões para ligação dos primers de sequenciamento Read1 e Read2.
Para a tarefa de ligação do adaptador, Ligase de DNA T4 (Cat#12996) é a escolha padrão. Ela exibe a capacidade de reparar cortes de fita simples encontrados em DNA fita dupla e reconectar nucleotídeos adjacentes.
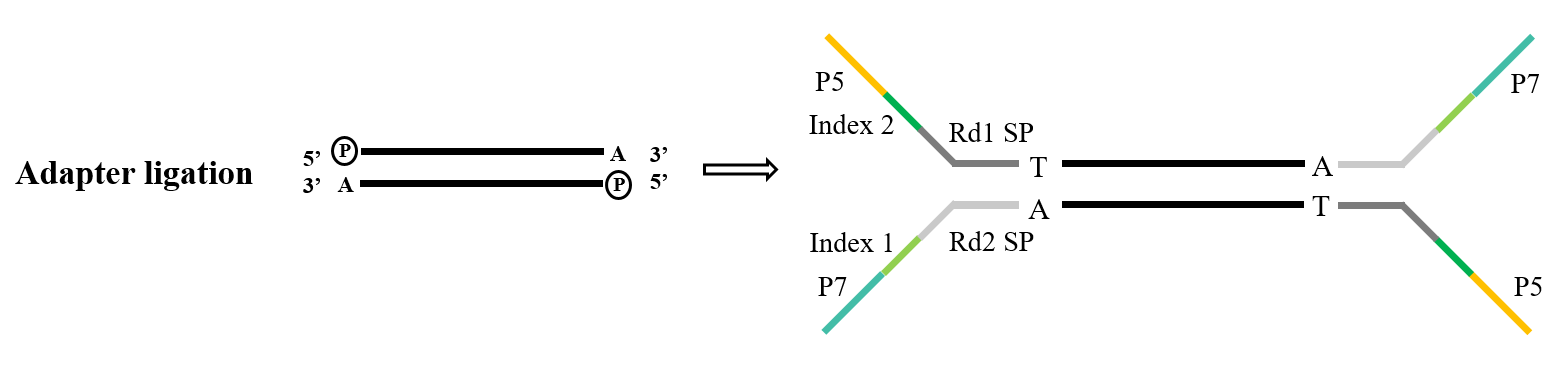
Figura 5. Processo geral de ligação do adaptador (Illumina)

Figura 6. Verificação de mutantes da ligase de DNA T4 pela ligação de DNA de 170 pb com adaptadores de 80 pb.
1.4 Amplificação por PCR
Obtenha sequências de DNA suficientes com adaptadores por meio da reação de PCR e conclua o sequenciamento da sequência de ácido nucleico da amostra na máquina. Hieff CanaceTM Pro High-Fidelity DNA Polymerase (Cat#13476) comumente usada em PCR tem atividade de polimerase 5'→3' e pode sintetizar DNA na direção 5'→3'. Além disso, também tem atividade de exonuclease 3'→5', que pode corrigir a incorporação errada de bases durante o processo de amplificação, para amplificar fragmentos de DNA rapidamente e com alta fidelidade.
2. Construção da biblioteca de RNA e suas principais enzimas
De acordo com os tipos de RNA, a construção de uma biblioteca de RNA pode ser dividida em biblioteca de mRNA, biblioteca de LncRNA, etc. A biblioteca de RNA convencional inclui os seguintes processos:
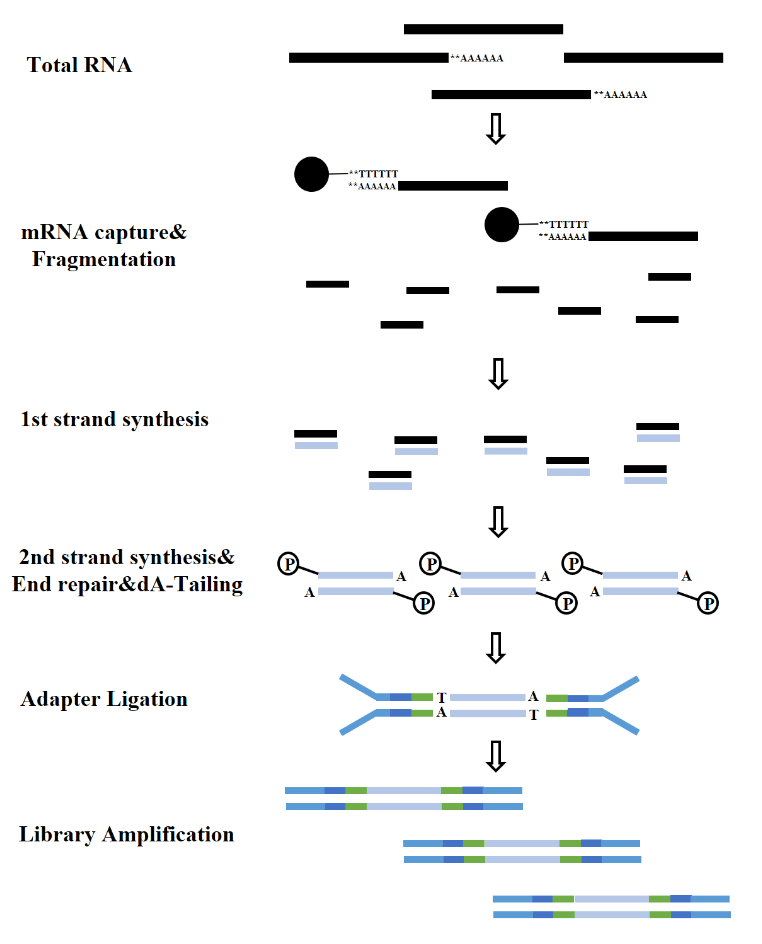
Figura 7. Processo de construção da biblioteca de mRNA (Illumina)
2.1 Enriquecimento de RNA
Seja lidando com eucariotos ou procariotos, o RNA ribossômico (rRNA) se destaca como o RNA mais abundante, constituindo até 80% do conteúdo total de RNA. Ao sequenciar o RNA total de uma amostra diretamente, uma porção substancial dos dados de sequenciamento estará relacionada ao rRNA. Para mitigar essa interferência, o método de enriquecimento de RNA deve ser empregado. Existem dois métodos principais para isso: enriquecimento de mRNA com base em métodos de oligo-dT e depleção de rRNA.
Em eucariotos, o mRNA exibe uma estrutura distinta de poli(A) na extremidade 3'. As esferas de Oligo-dT podem ser empregadas para capturar todo o mRNA transcrito da amostra, tornando-o adequado para análise transcricional, especialmente com amostras de RNA de alta qualidade. Por outro lado, os métodos de depleção de rRNA têm requisitos mais brandos sobre a qualidade da amostra e podem ser aplicados tanto a amostras de baixa qualidade (por exemplo, amostras FFPE) quanto a amostras de RNA de alta qualidade, bem como a amostras procarióticas. A abordagem comercial comumente usada envolve o uso da digestão com RNase H para remover rRNA, seguindo estas etapas específicas:
- Sintetizar sondas oligonucleotídicas específicas projetadas para se ligar ao rRNA.
- Empregue a RNase H (Cat#12906), que é capaz de degradar o RNA na fita híbrida RNA-DNA, para remover seletivamente o rRNA ligado às sondas.
- Por fim, digira as sondas de DNA com DNase I (Cat#10325), que pode degradar DNA de fita simples e dupla, eliminando efetivamente o rRNA. Para mais informações sobre DNase I, você pode seguir este link.
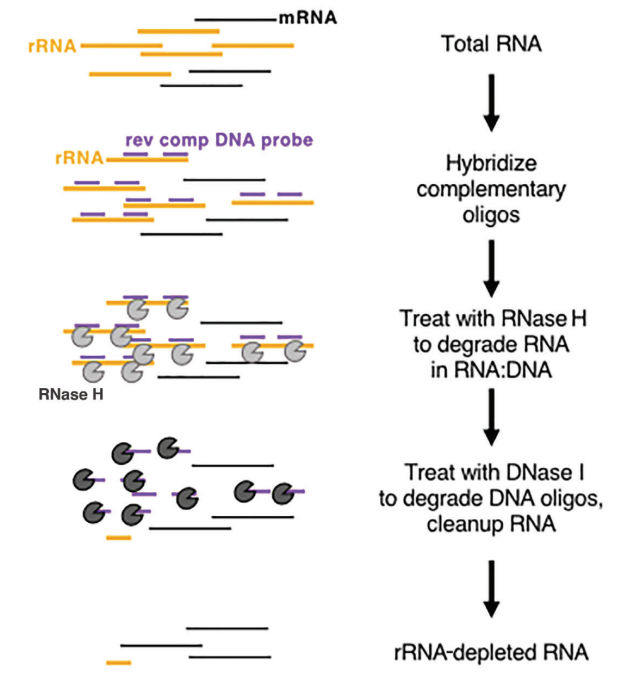
Figura 8: Diagrama esquemático da depleção de rRNA baseada em enzimas[5]
2.2 Fragmentação de RNA
Normalmente, sob a ação de cátions metálicos divalentes e altas temperaturas, grandes fragmentos de RNA são quebrados em pequenos fragmentos.
2.3 Síntese de cDNA de 1ª fita
Transcrição reversa do RNA alvo obtido na primeira fita de cDNA. Como o RNA é facilmente degradado por RNases presentes no ambiente, o uso de Inibidor de RNase (Cat#14672) durante a transcrição reversa pode inibir a atividade dessas enzimas e proteger o RNA da degradação da RNase. Ao mesmo tempo, transcriptase reversa (Cat#11112) foi usado para transcrever reversamente o RNA molde em cDNA. A transcriptase reversa tem atividade de DNA polimerase dependente de RNA e pode usar RNA como molde para sintetizar um cDNA na direção 5'→3'. A fita simples de DNA é complementar ao molde de RNA.
Durante o 1º síntese de cDNA de fita, a incorporação de actinomicina D melhorou inegavelmente a construção de bibliotecas específicas de fita, aumentando significativamente a especificidade da cadeia. Essa inovação agilizou o processo experimental, simplificando-o para os pesquisadores.
No entanto, a actinomicina D tem suas desvantagens: ela exibe toxicidade e requer proteção contra a luz. No cenário atual de crescente demanda por kits de construção de bibliotecas de placas e pré-misturados, a necessidade de proteção contra a luz impõe limitações aos avanços dos kits de placas.
Felizmente, a plataforma

2.4 Síntese de cDNA de 2ª fita
O cDNA de fita simples produzido por meio da transcrição reversa é altamente instável, necessitando da síntese imediata da segunda fita de cDNA sob a influência da DNA polimerase I. Durante essa síntese da segunda fita, a RNase H entra em ação removendo a fita de RNA da estrutura híbrida RNA-DNA. Ela funciona em conjunto com DNA polimerase I (Cat#12903) para facilitar a síntese catalítica da segunda fita complementar de cDNA. A DNA polimerase I possui atividade de DNA polimerase 5'→3' e, guiada por um molde e primer, sintetiza uma sequência que complementa o cDNA de fita simples na direção 5'→3'.
As etapas subsequentes no processo incluem reparo de extremidade, dA-Tailing, ligação do adaptador e amplificação por PCR, todas detalhadas no procedimento de construção da biblioteca de DNA e não precisam ser reiteradas aqui. Vale a pena notar que, uma vez que a transcrição reversa é concluída, não há necessidade de mais fragmentação do fragmento de ácido nucleico.
3. Diretriz para enzimas centrais NGS na construção de bibliotecas de DNA e RNA
A
Tabela 1.Diretriz para enzimas centrais NGS na construção de bibliotecas de DNA e RNA
| Tipo | Posicionamento do produto | Nome do produto | Gato# |
| Biblioteca de RNA construção | rRNA depleção/síntese de cDNA de 2ª fita | 12906ES | |
| rRNA esgotamento | 10325ES | ||
| Síntese de cDNA de 1ª fita | 14672ES | ||
| 11112ES | |||
| Síntese de cDNA de 2ª fita | 12903ES | ||
| Biblioteca de RNA construção & ADN biblioteca construção | Fim do reparo | 12901ES | |
| 12902ES | |||
| dA-Reboque | 13486ES | ||
| Ligadura adaptadora | 10301ES | ||
| PCR amplificação | 2×Super Canace® II Mix de alta fidelidade para amplificação de biblioteca | 12621ES |
Tabela 2.Biblioteca de DNA e RNA Kit de preparação
| Nome | Gato# | Notas | |
| ADN | Kit de preparação da biblioteca de DNA Hieff NGS | 13577ES | Tumor/ Método mecânico |
| Kit de preparação de biblioteca de DNA Hieff NGS OnePot Pro V2 | 12194ES | Tumor/ Método enzimático | |
| Hieff NGS OnePot II Kit de preparação de biblioteca de DNA para Illumina | 13490ES | Patógeno/ Enzimático/ tempo regular (140min) | |
| Kit de preparação de biblioteca de DNA Flash Hieff NGS OnePot | 12316ES | Patógeno/ Enzimático/ Ultra-rápido (100 minutos) | |
| Kit de co-preparação da biblioteca Hieff NGS DNA&RNA V2 | 12305ES | Co-preparação de patógenos/enzimáticos/DNA e RNA | |
| ARN | Kit de preparação de biblioteca de mRNA de modo duplo Hieff NGS Ultima | 12308ES | Sem esferas magnéticas oligo dT, 11 tubos |
| Kit de preparação de biblioteca de mRNA de modo duplo Hieff NGS Ultima | 12309ES | esferas magnéticas oligo dT plus, 14 tubos | |
| Kit de preparação de biblioteca de RNA de modo duplo Hieff NGS® Ultima | 12310ES | Versão pré-misturada, 5 tubos | |
| Hieff NGS ® EvoMax RNA Library Prep Kit (versão pré-misturada) (actinomicina D Livre) | 12340ES | Versão pré-misturada, (Actinomicina D Livre) | |
| Kit de depleção de rRNA Hieff NGS® MaxUp (planta) | 12254ES | Plantar | |
| Kit de depleção de rRNA humano Hieff NGS® MaxUp (rRNA e ITS/ETS) | 12257ES | Humano |
Referências:
[1] Mardis, Elaine R. Plataformas de sequenciamento de próxima geração[J]. Revisão Anual de Química Analítica, 2013, 6(1):287-303.
[2] Gulilat M, Lamb T, Teft WA, et al. Sequenciamento de próxima geração direcionado como uma ferramenta para medicina de recisão[J]. BMC Medical Genomics, 2019, 12(1):81.
[3] Lundberg KS, Dan DS, Adams M, et al. Amplificação de alta fidelidade usando uma DNA polimerase termoestável isolada de Pyrococcus furiosus[J]. Gene, 1991, 108(1):1-6.
[4] Miyazaki K. Fragmentação aleatória de DNA com endonuclease V: aplicação ao embaralhamento de DNA[J]. Nucleic Acids Research, 2002, 30(24):e139.
[5] Baldwin A, Morris AR, Mukherjee N. Um método fácil, econômico e escalonável para esgotar o RNA ribossômico humano para RNA-seq[J]. Protocolos atuais, 2021, 1(6):e176.