Die Hochdurchsatzsequenzierung, oft als Next-Generation-Sequencing-Technologie (NGS) bezeichnet, stellt einen bedeutenden Fortschritt gegenüber den ursprünglichen DNA-Sequenzierungsmethoden wie der Sanger-Sequenzierung dar. NGS ermöglicht die gleichzeitige Profilierung von Hunderttausenden, wenn nicht Millionen von Nukleinsäuremolekülsequenzen. Zu seinen Vorteilen zählen außergewöhnlicher Durchsatz, Kosteneffizienz, Skalierbarkeit und ein breites Anwendungsspektrum, was es zur weltweit vorherrschenden Sequenzierungstechnologie macht.
Der NGS-Sequenzierungsablauf umfasst vier Hauptphasen: Probenvorbereitung, Bibliotheksaufbau, Sequenzierung und Datenanalyse. Zentral für den Bibliotheksaufbau ist die Anfügung standardisierter NGS-Plattformadaptersequenzen an beide Enden fragmentierter genomischer DNA. Dieser Schritt zielt darauf ab, einen ausreichenden Vorrat an Bibliotheksnukleinsäuremolekülen zu erzeugen, die für die Sequenzierung auf dem NGS-Instrument durch PCR-Amplifikation vorbereitet sind. Je nach Art der Probe kann der NGS-Bibliotheksaufbau in DNA-Bibliotheksaufbau und RNA-Bibliotheksaufbau unterteilt werden. Enzyme spielen in diesen miteinander verbundenen Experimenten eine entscheidende Rolle. Welche Schlüsselenzyme sind also am Prozess des Bibliotheksaufbaus beteiligt?
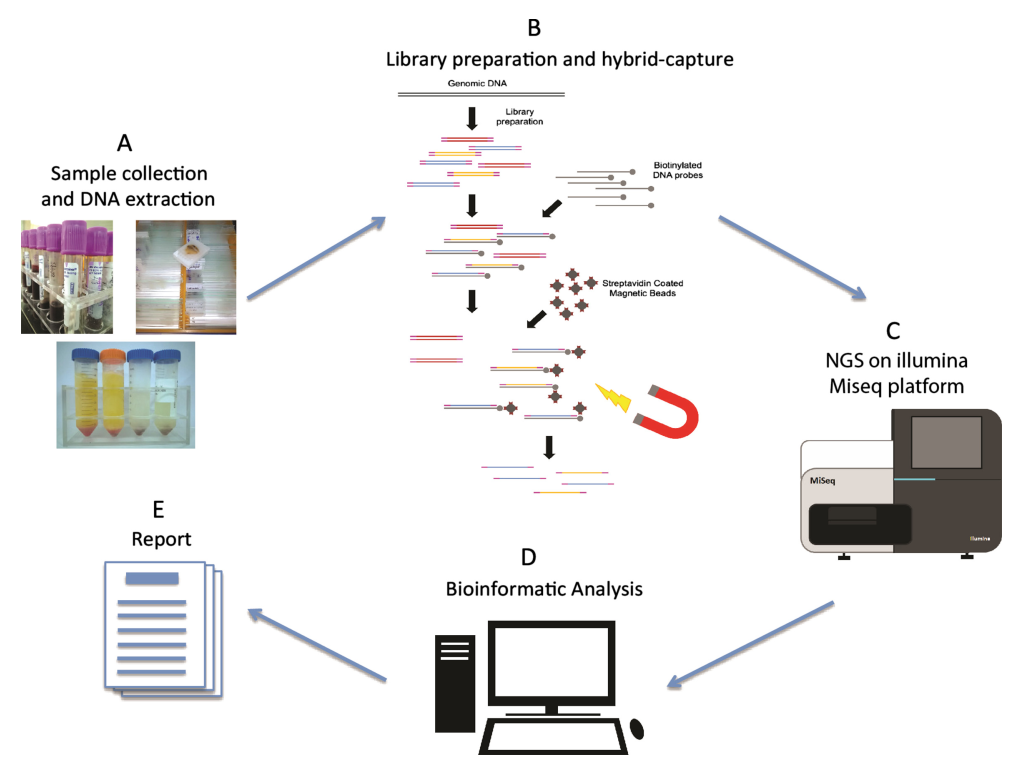
Abbildung 1. Sequenzierungsworkflow der nächsten Generation[2]
1. Aufbau einer DNA-Bibliothek und ihre Schlüsselenzyme
2. Aufbau einer RNA-Bibliothek und ihre Schlüsselenzyme
3. Leitfaden für NGS-Kernenzyme beim Aufbau von DNA- und RNA-Bibliotheken
1. Aufbau einer DNA-Bibliothek und ihre Schlüsselenzyme
Beim Aufbau einer DNA-Bibliothek ist der Aufbau einer TA-Klonligationsadapter-Bibliothek derzeit das am häufigsten verwendete technologische Mittel. Der Hauptprozess des Bibliotheksaufbaus ist wie folgt:

Abbildung 2. Prozess der DNA-Bibliothekskonstruktion (Illumina)
1.1 DNA-Fragmentierung
Die aktuellen Sequenzierer haben typischerweise eine Sequenzierungslänge im Bereich von 150-500 Basenpaaren (bp). Daher ist es notwendig, mechanische oder enzymatische Fragmentierungsmethoden anzuwenden, um große genomische DNA-Fragmente in kleinere zu zerlegen. Mechanische Fragmentierung kann zu relativ hohen Probenverlusten führen und ist mit einem komplizierteren Arbeitsablauf verbunden. Andererseits ist die enzymatische Verdauung eine häufig verwendete Methode zur Fragmentierung genomischer DNA. Im Vergleich zu mechanischen Methoden ist die enzymatische Verdauung kostengünstiger und unkomplizierter, da die Reaktion nach Zugabe des Fragmentierungsenzyms nur eine festgelegte Zeitspanne benötigt.
Derzeit werden hauptsächlich zwei Arten von Fragmenten verwendet. Eines basiert auf der Tn5-Transposase, die auf Transposonprinzipien basiert, während das andere eine Mischung aus Endonukleasen verwendet. Die Wirksamkeit dieser Fragmente kann jedoch durch den GC-Gehalt und die Basenpräferenzen der DNA beeinflusst werden. Im Gegensatz dazu bieten die von
1.2 Endreparatur, dA-Tailing
Die fragmentierte DNA erzeugt 5'/3'-Klebeenden und DNA mit stumpfen Enden. Alle Klebeenden müssen in stumpfe Enden umgewandelt werden, einschließlich der Entfernung von 3'-Überhängen und dem Auffüllen von 5'-überstehenden DNA-Enden. Wenn TA-Ligation für die Adapterligation verwendet wird, muss das DNA-Fragment auch am 5'-Ende phosphoryliert werden und am 3'-Ende muss ein „A“ hinzugefügt werden, um komplementär zum Adapter mit dem „T“-Klebeende zu sein.Der obige Prozess wird durch die Zusammenarbeit von T4-DNA-Polymerase, T4-Polynukleotidkinase und Taq DNA-Polymerase.
T4 DNA-Polymerase (Kat.-Nr. 12901) hat 5'→3' DNA-Polymerase-Aktivität, die die Synthese von DNA entlang der 5'→3'-Richtung katalysieren und das 5'-überstehende Ende auffüllen kann. Gleichzeitig hat das Enzym auch 3'→5' Exonuklease-Aktivität, um 3'-überhängende Enden abzuspalten und so DNA-Fragmente mit klebrigen Enden in DNA mit stumpfen Enden umzuwandeln.
Da die 5'-Enden synthetischer PCR-Primer und -Adapter normalerweise Hydroxylgruppen statt Phosphatgruppen sind, wird T4-Polynukleotidkinase (Kat.-Nr. 12902) benötigt, um die Übertragung von ATP-γ-Phosphatgruppen auf das 5'-Hydroxylende der Oligonukleotidkette in Gegenwart von ATP zu katalysieren und so den nächsten Schritt der Adapterligation vorzubereiten.
S-Taq DNA-Polymerase (Kat.-Nr. 13486) hat eine 5'→3'-Polymeraseaktivität, die DNA aus der 5'→3'-Richtung synthetisieren kann. Gleichzeitig hat es eine Desoxynucleotidyltransferaseaktivität, die ein Nukleotid „A“ an das 3'-Ende des PCR-Produkts anfügen kann.

Abbildung 3. Mehrere Enzyme sind am Endreparaturprozess beteiligt

Abbildung 4. S-taq fügt mit sehr hoher Effizienz A an die vier Basen von ATCG am 3'-Ende der durch Kapillarelektrophorese nachgewiesenen Gensegmente an.
1.3 Adapterligatur
Adapter stellen eine entscheidende Komponente der Bibliothek dar. Im Bereich der Illumina-Sequenzierung umfassen die häufig verwendeten Y-Typ-Adapter die Sequenzen P5/P7, Index und Rd1/Rd2 SP. Unter diesen dient die Sequenz P5/P7 dazu, sich mit der auf dem Sequenzierungschip vorhandenen Sequenz zu paaren und so die zu analysierenden Fragmente auf der Durchflusszelle zu verankern, um eine Brückenamplifikation durchzuführen. Die Indexsequenz wird verwendet, um zwischen verschiedenen Proben innerhalb der gemischten Bibliothek zu unterscheiden, die einer Sequenzierung unterzogen wird, während Rd1/Rd2 SP die Regionen zum Binden der Sequenzierungsprimer Read1 und Read2 bezeichnen.
Für die Aufgabe der Adapterligatur T4 DNA-Ligase (Kat.-Nr. 12996) ist die Standardauswahl. Es weist die Fähigkeit auf, einzelsträngige Bruchstellen in doppelsträngiger DNA zu reparieren und benachbarte Nukleotide wieder zu verbinden.
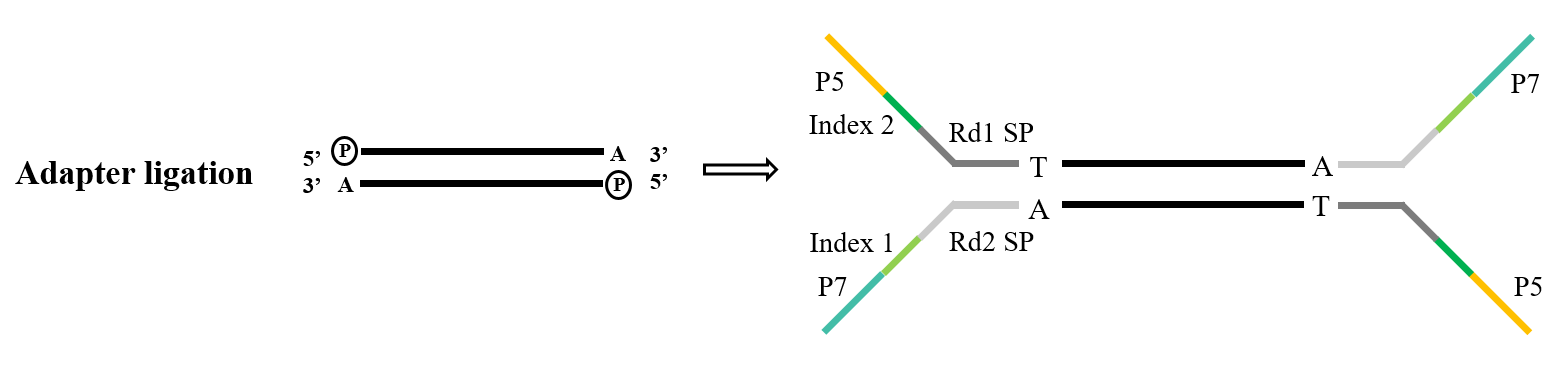
Abbildung 5. Allgemeiner Adapterligaturprozess (Illumina)

Abbildung 6. Überprüfung von T4-DNA-Ligase-Mutanten durch Ligation von 170-bp-DNA mit 80-bp-Adaptern.
1.4 PCR-Amplifikation
Erhalten Sie mithilfe von Adaptern durch eine PCR-Reaktion genügend DNA-Sequenzen und schließen Sie die Sequenzierung der Nukleinsäuresequenz der Probe auf dem Gerät ab. Hieff CanaceTM Die häufig bei der PCR verwendete Pro High-Fidelity DNA-Polymerase (Kat.-Nr. 13476) hat eine 5'→3'-Polymeraseaktivität und kann DNA in 5'→3'-Richtung synthetisieren. Darüber hinaus besitzt sie auch die Aktivität einer 3'→5'-Exonuklease, die den falschen Einbau von Basen während des Amplifikationsprozesses korrigieren kann, um DNA-Fragmente schnell und mit hoher Genauigkeit zu amplifizieren.
2. Aufbau einer RNA-Bibliothek und ihre Schlüsselenzyme
Je nach RNA-Typ kann der Aufbau einer RNA-Bibliothek in mRNA-Bibliothek, LncRNA-Bibliothek usw. unterteilt werden. Die herkömmliche RNA-Bibliothek umfasst die folgenden Prozesse:
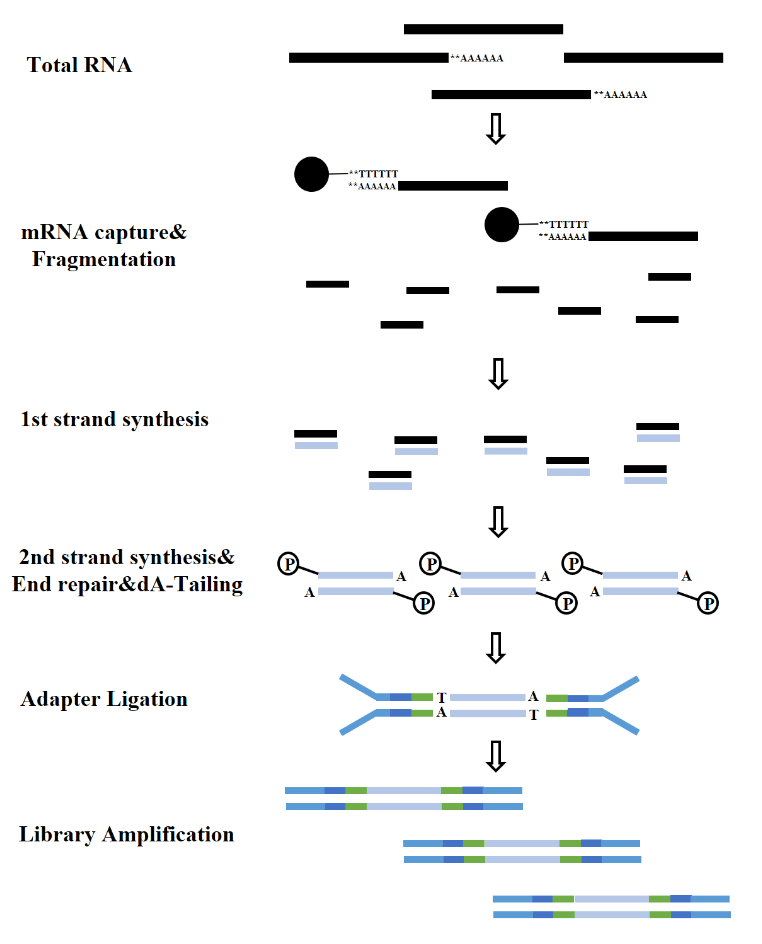
Abbildung 7. Prozess der mRNA-Bibliothekskonstruktion (Illumina)
2.1 RNA-Anreicherung
Ob bei Eukaryoten oder Prokaryoten, ribosomale RNA (rRNA) ist die am häufigsten vorkommende RNA und macht bis zu 80 % des gesamten RNA-Gehalts aus. Bei der direkten Sequenzierung der gesamten RNA einer Probe wird ein erheblicher Teil der Sequenzierungsdaten mit rRNA in Zusammenhang stehen. Um diese Interferenz zu mildern, muss die Methode der RNA-Anreicherung eingesetzt werden. Dafür gibt es zwei Hauptmethoden: mRNA-Anreicherung basierend auf Oligo-dT und rRNA-Depletionsmethoden.
Bei Eukaryoten weist mRNA am 3'-Ende eine ausgeprägte Poly(A)-Struktur auf. Oligo-dT-Perlen können verwendet werden, um die gesamte aus der Probe transkribierte mRNA zu erfassen, was sie für die Transkriptionsanalyse geeignet macht, insbesondere bei hochwertigen RNA-Proben. rRNA-Depletionsmethoden hingegen stellen geringere Anforderungen an die Probenqualität und können sowohl auf Proben geringer Qualität (z. B. FFPE-Proben) als auch auf RNA-Proben hoher Qualität sowie auf prokaryotische Proben angewendet werden. Der häufig verwendete kommerzielle Ansatz beinhaltet die Verwendung von RNase H-Verdauung zur Entfernung von rRNA, wobei diese spezifischen Schritte befolgt werden:
- Synthetisieren Sie spezifische Oligonukleotidsonden, die für die Bindung an rRNA entwickelt wurden.
- Verwenden Sie RNase H (Kat.-Nr. 12906), die RNA im RNA-DNA-Hybridstrang abbauen kann, um die an die Sonden gebundene rRNA selektiv zu entfernen.
- Zum Schluss verdauen Sie die DNA-Sonden mit DNase I (Kat.-Nr. 10325), das sowohl einzelsträngige als auch doppelsträngige DNA abbauen kann und rRNA effektiv eliminiert. Weitere Informationen zu DNase I finden Sie unter diesem Link.
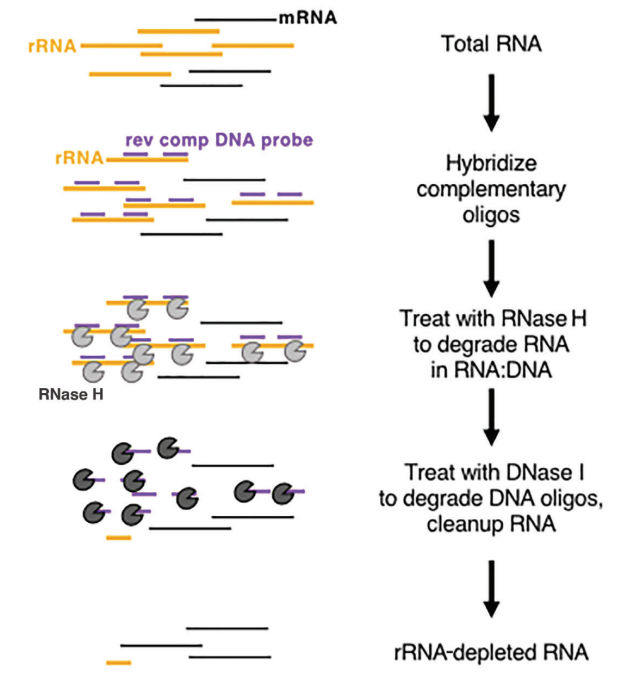
Abbildung 8: Schematische Darstellung der enzymatischen rRNA-Depletion[5]
2.2 RNA-Fragmentierung
Normalerweise werden große RNA-Fragmente unter der Einwirkung zweiwertiger Metallkationen und hoher Temperaturen in kleine Fragmente zerlegt.
2.3 Synthese des 1. Strangs der cDNA
Reverse Transkription der erhaltenen Ziel-RNA in den ersten Strang der cDNA. Da RNA leicht durch in der Umwelt vorhandene RNasen abgebaut wird, ist die Verwendung von RNase-Inhibitor (Kat.-Nr. 14672) während der reversen Transkription kann die Aktivität dieser Enzyme hemmen und RNA vor RNase-Abbau schützen. Gleichzeitig Reverse Transkriptase (Kat.-Nr. 11112) wurde verwendet, um die Matrizen-RNA in cDNA umzuwandeln. Die Reverse Transkriptase hat eine RNA-abhängige DNA-Polymerase-Aktivität und kann RNA als Matrizen verwenden, um eine cDNA in 5'→3'-Richtung zu synthetisieren. Der DNA-Einzelstrang ist komplementär zur RNA-Matrize.
Während des 1. Die Einbeziehung von Actinomycin D hat bei der Synthese von Strang-cDNA die Konstruktion strangspezifischer Bibliotheken unbestreitbar verbessert und die Kettenspezifität deutlich erhöht. Diese Innovation hat den experimentellen Prozess rationalisiert und ihn für Forscher vereinfacht.
Actinomycin D hat jedoch auch seine Nachteile: Es ist toxisch und muss vor Licht geschützt werden. Angesichts der steigenden Nachfrage nach vorgefertigten Kits und Kits zum Aufbau von Plattenbibliotheken stellt die Notwendigkeit des Lichtschutzes eine Einschränkung für die Weiterentwicklung von Plattenkits dar.
Glücklicherweise hat die

Abbildung 9: Engineering von MMLV zur Identifizierung von MMLV-Mutanten, die zur Standard-RNA-Sequenzierung beitragen könnten
2.4 Synthese des zweiten cDNA-Strangs
Die durch Reverse Transkription produzierte einzelsträngige cDNA ist höchst instabil, was die sofortige Synthese des zweiten cDNA-Strangs unter dem Einfluss der DNA-Polymerase I erforderlich macht. Während dieser Zweitstrangsynthese kommt RNase H ins Spiel, indem sie den RNA-Strang aus der RNA-DNA-Hybridstruktur entfernt. Sie arbeitet zusammen mit DNA-Polymerase I (Kat.-Nr. 12903) um die katalytische Synthese des komplementären zweiten Strangs der cDNA zu erleichtern. DNA-Polymerase I besitzt 5'→3'-DNA-Polymeraseaktivität und synthetisiert, geleitet von einer Vorlage und einem Primer, eine Sequenz, die die einzelsträngige cDNA in der 5'→3'-Richtung ergänzt.
Die nachfolgenden Schritte im Prozess umfassen Endreparatur, dA-Tailing, Adapterligation und PCR-Amplifikation, die alle im Verfahren zur DNA-Bibliothekserstellung ausführlich beschrieben sind und hier nicht wiederholt werden müssen. Es ist erwähnenswert, dass nach Abschluss der Reverse-Transkription keine weitere Fragmentierung des Nukleinsäurefragments erforderlich ist.
3. Leitfaden für NGS-Kernenzyme beim Aufbau von DNA- und RNA-Bibliotheken
Tabelle 1.Leitfaden für NGS-Kernenzyme beim Aufbau von DNA- und RNA-Bibliotheken
| Typ | Produktpositionierung | Produktname | Katze# |
| RNA-Bibliothek Konstruktion | rRNA Depletion/2. Strang cDNA Synthese | 12906ES | |
| rRNA Erschöpfung | 10325ES | ||
| 1. Strang cDNA Synthese | 14672ES | ||
| 11112ES | |||
| Synthese der 2. cDNA-Strang | 12903ES | ||
| RNA-Bibliothek Konstruktion und DNA Bibliothek Konstruktion | Reparatur beenden | 12901ES | |
| 12902ES | |||
| dA-Tailing | 13486ES | ||
| Adapterligatur | 10301ES | ||
| PCR Verstärkung | 2×Super Canace® II High-Fidelity-Mix zur Bibliotheksverstärkung | 12621ES |
Tabelle 2.DNA- und RNA-Bibliothek Vorbereitungskit
| Name | Katze# | Hinweise | |
| DNA | Hieff NGS DNA-Bibliotheksvorbereitungskit | 13577ES | Tumor/ Mechanische Methode |
| Hieff NGS OnePot Pro DNA-Bibliotheksvorbereitungskit V2 | 12194ES | Tumor/ Enzymatische Methode | |
| Hieff NGS OnePot II DNA Library Prep Kit für Illumina | 13490ES | Pathogen/ Enzymetisch/ normale Zeit (140min) | |
| Hieff NGS OnePot Flash DNA-Bibliotheksvorbereitungskit | 12316ES | Pathogen/ Enzymatisch/ Ultraschnell (100 Minuten) | |
| Hieff NGS DNA&RNA Library Co-Prep Kit V2 | 12305ES | Pathogen/ Enzyme/ DNA & RNA Co-Prep | |
| RNA | Hieff NGS Ultima Dual-Mode mRNA-Bibliotheksvorbereitungskit | 12308ES | Ohne Oligo dT Magnetkügelchen, 11 Röhrchen |
| Hieff NGS Ultima Dual-Mode mRNA-Bibliotheksvorbereitungskit | 12309ES | Oligo dT Magnetperlen plus, 14 Röhrchen | |
| Hieff NGS® Ultima Dual-Mode RNA-Bibliotheksvorbereitungskit | 12310ES | Vorgemischte Version, 5 Tuben | |
| Hieff NGS ® EvoMax RNA Library Prep Kit (Vorgemischte Version) (Actinomycin D Frei) | 12340ES | Vorgemischte Version, (Actinomycin D Frei) | |
| Hieff NGS® MaxUp rRNA-Depletion Kit (Pflanze) | 12254ES | Anlage | |
| Hieff NGS® MaxUp Human rRNA Depletion Kit (rRNA und ITS/ETS) | 12257ES | Menschlich |
Verweise:
[1] Mardis, Elaine R. Sequenzierungsplattformen der nächsten Generation[J].
[2] Gulilat M, Lamb T, Teft WA, et al. Gezieltes Next-Generation-Sequencing als Instrument der Resektionsmedizin[J]. BMC Medical Genomics, 2019, 12(1):81.
[3] Lundberg KS, Dan DS, Adams M, et al. Hochpräzise Amplifikation mithilfe einer thermostabilen DNA-Polymerase, isoliert aus Pyrococcus furiosus[J]. Gene, 1991, 108(1):1-6.
[4] Miyazaki K. Zufällige DNA-Fragmentierung mit Endonuklease V: Anwendung für das DNA-Shuffling[J]. Nucleic Acids Research, 2002, 30(24):e139.
[5] Baldwin A, Morris AR, Mukherjee N. Eine einfache, kostengünstige und skalierbare Methode zur Depletion menschlicher ribosomaler RNA für RNA-seq[J]. Current Protocols, 2021, 1(6):e176.